The human body is inhabited by around trillions of microbes composing a multicomplex system, termed microbiota, which is strongly involved in the regulation and maintenance of homeostasis. Perturbations in microbiota composition can lead to dysbiosis, which has been associated with several human pathologies. The gold-standard method to explore microbial composition is next-generation sequencing, which involves the analysis of 16S rRNA, an indicator of the presence of specific microorganisms and the principal tool used in bacterial taxonomic classification. Indeed, the development of 16S RNA sequencing allows us to explore microbial composition within the human body, including fluids, since it has been detected in “germ-free” environments such as blood, plasma, and urine of diseased and healthy subjects. Recently, prokaryotes showed to generate extracellular vesicles, which are known to be responsible for shuttling different intracellular components such as proteins and nucleic acids (including 16S molecules) by protecting their cargo from degradation. These vesicles can be found in several human biofluids and can be exploited as tools for bacterial detection and identification.
- 16S
- plasma
- extracellular vesicles
- microbiome
Note:Dear author, the following contents are excerpts from your papers. They are editable. And the entry will be online only after authors edit and submit it.
1. The Microbiome as a Gateway to Detect the Microbiota Composition: 16S Sequencing
The constant increase in the number of studies on the microbiota and its interactions with the human organism is largely due to the use of new technologies involving sequencing analysis. In fact, before the adoption of next-generation sequencing (NGS) in this field, the detection of microorganisms from different human districts was conducted by culturing methods, but, beyond being time-consuming, these tools were greatly hampered and unsuccessful due to “unculturable” bacteria [6].
In time, it became evident for the necessity to identify markers allowing for the detection of microorganisms in a particular substrate and, together, their identification at the species level with high confidence. A very good response to this unmet need was represented by a specific class of RNAs. In bacteria, the 5S, 16S, and 23S ribosomal RNAs are organized in a gene cluster which is expressed as a single operon. The size, sequence, and secondary structures of these three rRNA genes are highly conserved between different bacterial species. In particular, the 16S rRNA [7] originates from a gene of about 1500 bp present in all bacteria and contains nine hypervariable and species-specific regions (V1—V9) flanked by highly conserved and well-known portions of the genome. Thus, by using specific PCR primers, it is possible to amplify parts of the gene containing both the constant and the variable sequences, which are very useful for microbial classification (Figure 1). Indeed, this gene has been used for phylogenetic studies for many years and is considered the gold standard for microbiome detection and classification [8].
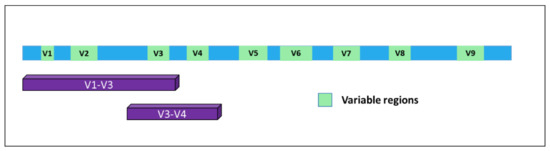
Figure 1. Structure of the 16S gene. The nine variable regions are depicted in green. Purple bars indicate the portions of the gene mostly used for bacterial classification upon PCR-based amplification and sequencing.
The first example of 16S amplification and sequencing using the Sanger method was presented in 1990 but was soon followed by the advent of high throughput sequencing [9,10], which represented a turning-point and a revolution in terms of costs and speed of detection, boosting the development of microbiome-based studies. Indeed, Sanger sequencing following PCR amplification was successful in identifying bacterial strains in monomicrobial infections, for example, outperforming conventional culture methods or diagnostic body fluid markers, but failed to identify unique molecular targets in polymicrobial infections. This is due to the intrinsic property of the Sanger method, which can be performed only on DNA molecules with the same sequence, while NGS allows for massive parallel sequencing of many different DNA templates.
In 2007, the Human Microbiome Project (HMP) was started to use next-generation sequencing methods to identify the abundance, diversity, and functionality of the microorganisms that live in different sites of the human body, thus generating a reference database for subsequent comparative analyses [11]. With this aim, 16S-focused investigations were implemented. The 16S analysis typically starts with the amplification of specific variable regions of the 16S rRNAs (usually V3—V4), to be massively sequenced in parallel; then, the obtained sequences are clustered into Operational Taxonomic Units (OTUs) [12]. These are defined according to their similarity to each other based on a threshold, usually defined as a sequence similarity of at least 97%. In time, several bioinformatics pipelines have been implemented to analyze the results of 16S sequencing, such as QIIME (Quantitative Insights Into Microbial Ecology, http://qiime.org/) [13] and MOTHUR (https://mothur.org/) [14], which were specifically designed for examining microbial communities.
The two principal parameters describing the complexity of microbiota in a definite environment are the α and β diversity. Alpha diversity describes the richness and evenness, i.e., the number of different organisms and the homogeneity of their distribution within a sample. Beta diversity is a measure of absolute or relative overlap in taxa shared between samples. There is a wide range of microbial β diversity in the microbiota between individuals since particular species could be widely abundant in some individuals and may be minimally represented in others [15]. In particular, more specific indexes belonging to both alpha and beta metrics are often used during the assessment and classification of bacterial communities. Among the most used there are the Shannon-Weaver, the Simpson, the Jaccard, and the Bray-Curtis, which quantify the taxonomic dissimilarity. Since all diversity indices have specific biases, they must be selected appropriately [16,17].
2. Circulating 16S in a Disease Context
As previously said, numerous studies analyzed the role of the microbiome in relation to the onset of different pathologies, most of them focusing on the intestinal microbiome. Lately, though, an increasing number of researchers investigating connections between dysbiosis in the blood microbiome and human disease. In a work from 2011, Amar et al. showed, for the first time, that the blood microbiome composition might predict the onset of diabetes, in a 6–9-year follow-up [41]. About a year later, the authors performed 16S qPCR on the blood of individuals without CVD at baseline and found a decrease in blood bacterial DNA and an increase of Proteobacteria in subjects who suffered from cardiovascular complications during the follow-up [42]. In a subsequent work in 2014 on circulating human microbiome in CVD subjects, Dinakaran et al. found an increase in microbial diversity and bacterial DNA concentration in the blood of patients [43]. Here, they observed a predominance of Actinobacteria, while the most abundant phylum in healthy subjects was Proteobacteria.
In a different study, Lelouvier et al. investigated the association between blood microbiota and the onset of liver disease [44]. The authors performed both 16S qPCR and sequencing to unveil the relationship between blood bacterial population composition and liver fibrosis in obese patients. The results showed higher concentrations of 16S in the blood of patients with fibrosis than in healthy subjects, thus identifying a specific microbial cluster associated with liver fibrosis that was suggested as a biomarker for its early detection.
Qian et al. in 2018 investigated the possible association between blood microbiota alteration and Parkinson’s disease [45]. The taxonomic diversity was assessed by performing 16S sequencing and some genera resulted associated with the pathology, with Cloacibacterium, Isoptericola, Paludibacter, and Saccharofermentans genus showing a correlation with disease duration.
More recently, a manuscript from Hammad and co-authors described the profiles of circulating microbial DNA in patients with rheumatoid arthritis (RA) in comparison with patients affected by ankylosing spondylitis (AS) or psoriatic arthritis (PA) and healthy control subjects [46]. Bacterial community members were identified by sequencing of the 16S rRNA variable region 4 in all samples. At the phylum level, the blood microbiome was predominated by four phyla, i.e., Proteobacteria, Firmicutes, Bacteroidetes and Actinobacteria, supporting the notion of a core blood microbiome as reported in previous publications [39,42,44,47]. In 2016, Santiago and coworkers presented an investigation assessing serum microbial composition in patients with and without ascites [48]. They performed 16S rDNA high-throughput sequencing, evidencing complex and specific microbial communities in serum and ascitic fluid of patients with cirrhosis. Interestingly, sera obtained from healthy controls resulted in an almost complete absence of bacteria. Of note, the authors indicated the presence of an unknown phylum belonging to Cyanobacteria in the serum of patients with ascites.
3. Source of Circulating 16S RNA
The source of circulating 16S molecules has been discussed for many years. Some researchers hypothesized that the bacteria in the blood originated from gastro-intestinal tract leakage, but it has also been suggested that they could derive from skin or the oral tract and that they diffuse in blood when these protective barriers are compromised [38]. An additional hypothesis, though, could be represented by extracellular bacterial vesicles. Indeed, both Gram-negative and Gram-positive bacteria can release spherical membrane vesicles (MV) derived by the cell membrane and containing several molecules involved in different functions [49]. To date, the studies on the biogenesis, structure, and function of MV in Gram-positive bacteria are just beginning, while detailed studies have been conducted on Gram-negative microbes.
To better understand the structure and function of these vesicles, it is worth making a brief introduction to the composition of the bacterial envelope. The cellular structure of Gram-negative bacteria is characterized by a double cell membrane divided by a periplasmic space containing a layer of peptidoglycan. The outer membrane (OM) is quite peculiar, as it is composed of an outer leaflet consisting of lipopolysaccharide (LPS) and an inner leaflet consisting of phospholipids. The presence and distribution of LPS have a tremendous impact on the ability of the bacteria to survive in harsh environments, as they regulate the impermeability to hydrophobic compounds such as antibiotics and detergents. Differently, the inner membrane is composed of a phospholipid bilayer and proteins and encloses the contents of the bacterial cell [50,51].
The outer membrane vesicles (OMV) are spherical structures of 20–300 nm produced by Gram-negative bacteria, and derive by the blebbing of the outer plasmatic membrane, being composed of the outside membrane and periplasmic material. The detachment of the outer membrane, in pathogenic and nonpathogenic bacteria, is not limited to a stochastic process of fragmentation and splitting but is a finely controlled process occurring during the normal growth of bacteria and in which several environmental factors are involved [52]. Different triggers of vesicle biogenesis were identified in Gram-negative bacteria, involving alteration in peptidoglycans structure, accumulation of LPS, and enrichment of the outer membrane with phospholipids. Moreover, several pieces of evidence showed that alteration of the microbial structure caused by various environmental factors, such as antibiotics or temperature variations, may cause vesicle accumulation [53].
Differently from Gram-negative bacteria, the biogenesis and composition of extracellular vesicles in Gram-positive bacteria remain quite elusive. The protein composition of extracellular vesicles (EVs) produced by the Gram-positive bacterium Staphylococcus aureus was investigated in 2009. EVs were reported to be similar in size to their Gram-negative counterparts (20–100 nm in diameter), while their cargo included a variety of proteins that are important for survival and virulence [54]. Since Gram-positive are characterized by a thick peptidoglycan cell wall outside of the cell membrane, the production and release of MV is a highly regulated and complex process. To date, three different mechanisms of EVs release have been proposed: (1) the vesicles may be pushed through the wall by turgor pressure, and their size could be affected by cell wall pore size or thickness; (2) cell wall modifications could be induced by specific modifying enzymes, thus facilitating its loosening and triggering EV release; (3) specific channel-like structures could facilitate EVs passage through pores, guided by tubulin [55].
Regardless of their source, the cargo of bacterial EVs displays quite a heterogeneous arrangement, including inner-membrane, periplasmic and cytoplasmic components, genetic material (DNA/RNA), toxins, and also factors involved in antibiotic resistance. OMVs are essential for different functions such as cell-to-cell communication, the formation of biofilms, bacterial infections, and the transfer of proteins and genetic material [56].
This entry is adapted from the peer-reviewed paper 10.3390/ijms21238959