Peritoneal carcinomatosis is a challenging condition that affects many cancer patients, and conventional therapies have limited efficacy in treating it. However, recent advances in the field of immunotherapy have shown promise in improving treatment outcomes. One promising approach is immune checkpoint inhibitors, which block proteins that inhibit T-cell activity and promote an anti-tumor immune response. Another approach involves the use of CAR-T cells, which are genetically modified T cells engineered to recognize and target cancer cells expressing specific antigens. In addition, dendritic cells and vaccine-based therapeutics are also designed to stimulate the immune system to recognize and attack cancer cells.
- intraperitoneal immunotherapy
- peritoneal carcinomatosis
- ascites
1. Introduction
2. Peritoneal Carcinomatosis
2.1. Peritoneum and Peritoneal Carcinomatosis
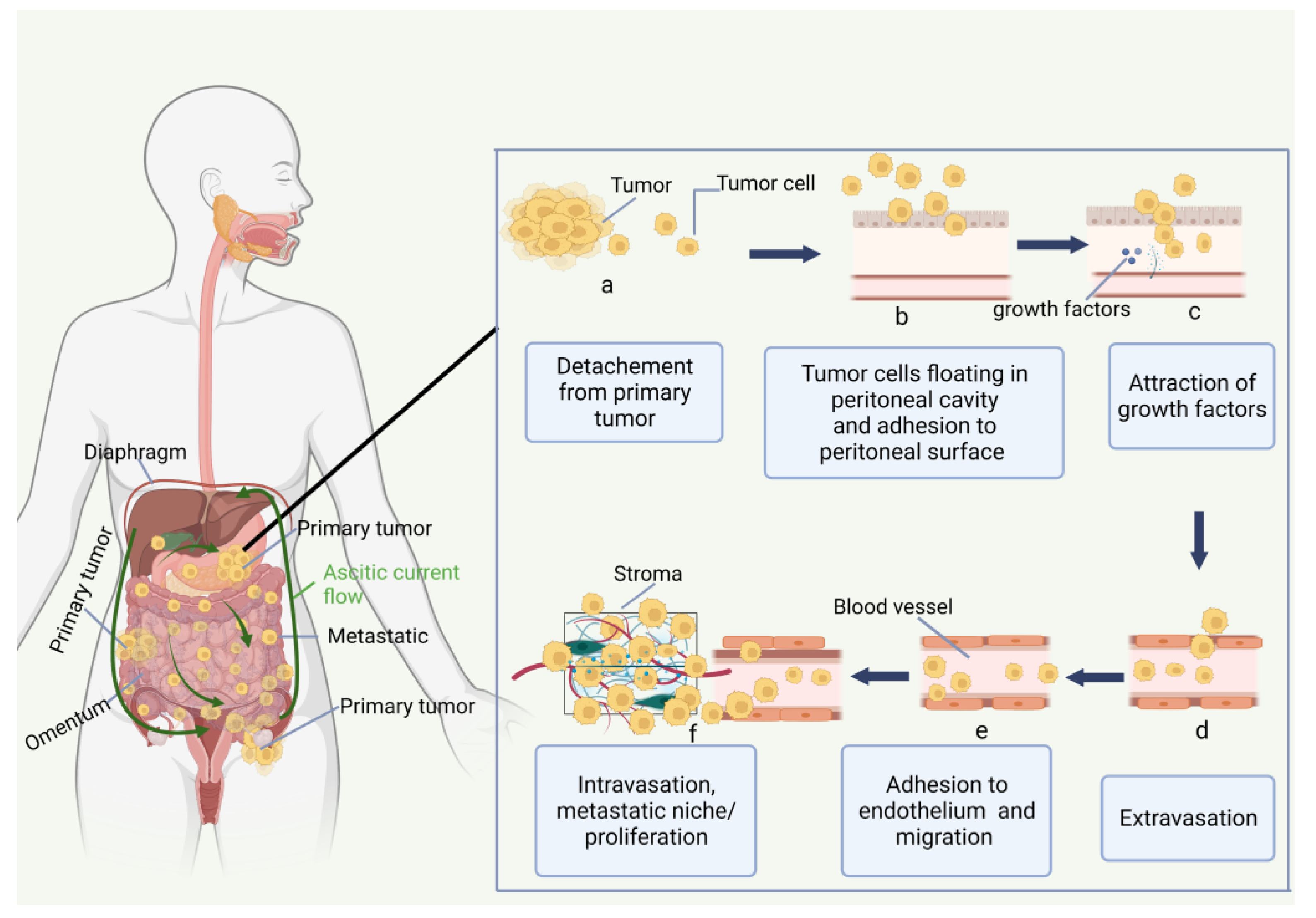
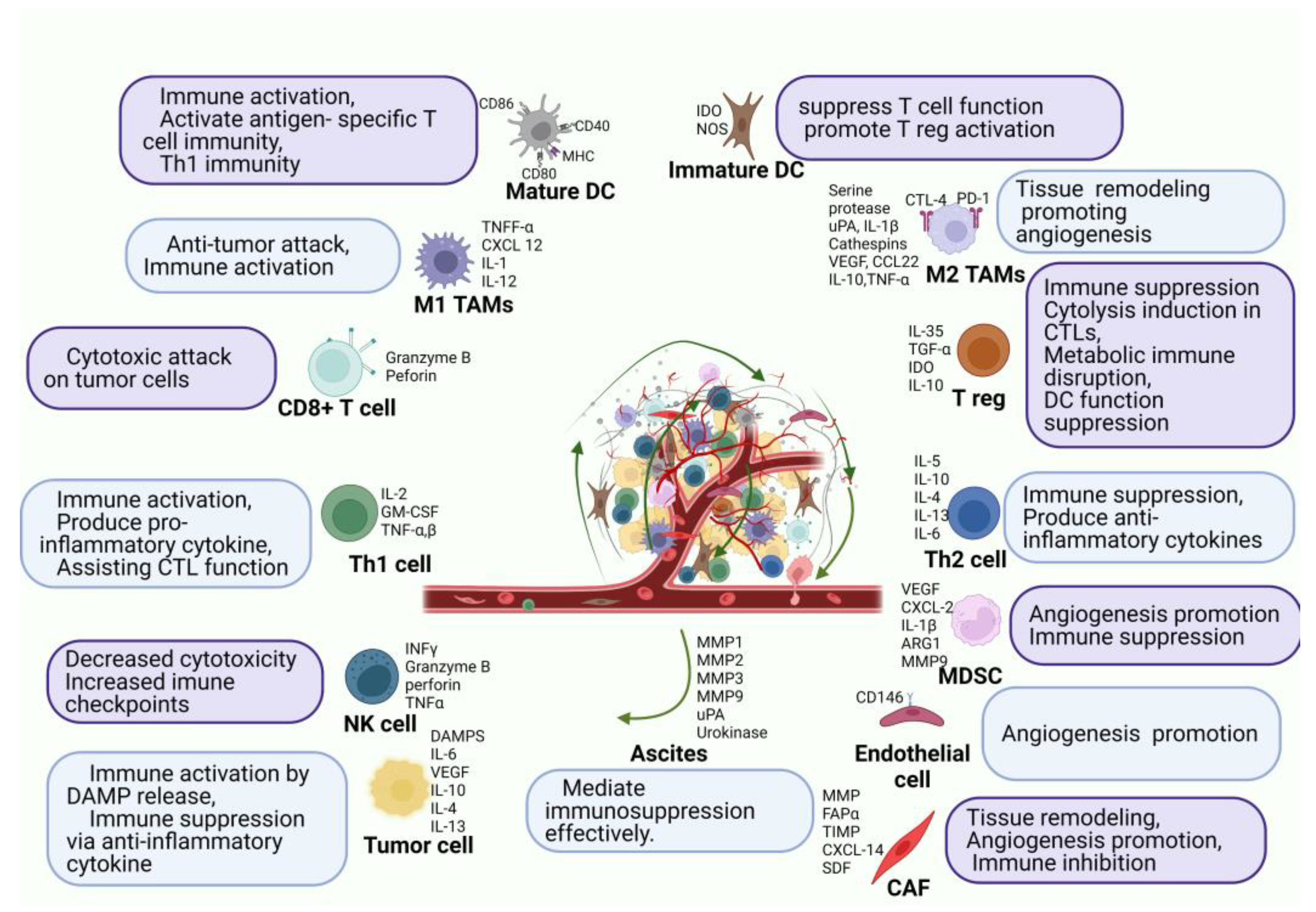
2.2. Immune Environment of Peritoneal Carcinomatosis
3. Immune Checkpoint Inhibitors
4. Monoclonal Antibodies
4.1. MOC31PE Immunotoxin
4.2. Catumaxomab
5. Cancer Vaccines for Peritoneal Metastasis
Therapeutic vaccines against cancer are a further immunotherapy strategy that has attracted substantial recent advancements in the intraperitoneal developments of PC. Malignant ascites have a bad prognosis and are a significant barrier to the immune system responding to vaccines. To combat this, vaccines are currently being developed and modified to specifically target ascites to enhance the quality of life for PM patients.
Cellular, viral vector, and molecular (peptide, DNA, or RNA) are the three main platforms for cancer vaccines [62]. Allogeneic tumor cell lines or autologous patient-derived tumor cells are used to create cellular vaccines [63]. Due to their functions as tumor antigen consumers, processors, and presenters, dendritic cells (DCs) are employed to create cellular cancer vaccines. Oncolytic viral vaccinations have been genetically altered to target and kill tumor cells [64]. In addition to their oncolytic effects, viral vectors also stimulate tumor-specific immune responses by providing tumor antigens through more typical T-cell priming procedures [65]. On the cell surface, major histocompatibility complex (MHC) peptides expression can be detected by T-cells [66]. For the creation of peptide-based cancer vaccines, it is important to know how peptides and T cell receptors interact with MHC. Enzymes break down short peptides, which are typically nine amino acid residues long, and immediately connect to MHC molecules, perhaps generating tolerance [67]. Longer peptides, typically 30 mer, are taken in by antigen-presenting cells (APCs), processed for MHC presentation, and result in memory CD4+ and CD8+ T cell immunological responses, which may make APCs more immunogenic [67]. DNA vaccines, often known as “naked DNA”, are closed circular DNA plasmids that encode TAAs and immunomodulatory substances intending to induce tumor-specific immune responses [68]. Despite being straightforward, secure, and quick to create, naked DNA vaccines are ineffective against target tumor cells due to low rates of transfection. mRNA vaccines, which are produced in vitro, encode an antigen or antigens, and following internalization, they express proteins that cause an immune reaction. mRNA vaccines may convey a large number of antigens and co-stimulatory signals without running the risk of infection or insertional mutagenesis, and their manufacture is rapid and affordable. However, the delivery effectiveness and stability are issues for mRNA vaccines [68].
Targeting ascites in PC has been accomplished by combining DCs with cytokine-induced killer cells (CIKs), which are cytotoxic T lymphocytes with a CD3+ CD56+ phenotype. The choice of CIKs was made based on three important criteria: they exhibit low cytotoxicity toward normal cells, no negative impact on hematopoiesis in the bone marrow, and resistance to Fas ligand-induced apoptosis. The effects of the combined treatment of DCs and CIKs include an increase in cytotoxic T cells in ascites that are driven by TNF and IFN and a reduction in immunosuppressive Tregs [69]. Similar to CAR-T cells, the method by which cancer vaccines are administered plays an important role in their dissemination. Natural killer cells (NKs) and dendritic cells (DCs) working together to fight tumors have been proven to be successful. Geller et al. demonstrated that IP- injection of IL-2-activated NK cells enhanced antitumor effects in an ovarian cancer mouse model xenograft, in contrast to systemic distribution [70].
Many cycles of patient-derived type I CD4+ T helper cells (Th1) provided by IP together with the cytokines IL-2 and IFN were shown to improve the anti-tumor activity of autologous CD8+ T cells against the tumor-specific glycoform of MUC1. This was reported by Dobrzanski et al. [71][72]. In a peritoneal metastatic colon cancer murine model, Alkayyal et al. further emphasized the relevance of combining pro-inflammatory cytokine IL-12 with an oncolytic virus (Maraba MG1) for reducing tumor burden in a CT26 colon cancer model. When MG1-IL12-ICV was IP-administered to these animals, it significantly decreased tumor development, created resistance to CT26 cell reinoculation, and improved survival. Regarding the mechanism, IL-12 was effective in enticing NK cells to the tumor location for annihilation. When paired with MG1 viral proteins, these activated NK cells generated IFN, which stimulated DCs and aided in the attraction of more NK cells [73].
6. CAR-T Cell Therapy for Peritoneal Carcinomatosis
6.1. Basic of CAR-T Cells
6.2. Administration Route of CAR-T for Peritoneal Carcinomas
6.3. CAR-T Cell Studies for Peritoneal Carcinomatosis
This entry is adapted from the peer-reviewed paper 10.3390/cancers15082383
References
- Willaims, S.C.P. Peritoneal Carcinomatosis: Cause, Symptoms, Diagnosis, and Treatment. Available online: https://www.webmd.com/cancer/what-is-peritoneal-carcinomatosis (accessed on 9 July 2022).
- Coccolini, F.; Gheza, F.; Lotti, M.; Virzì, S.; Iusco, D.; Ghermandi, C.; Melotti, R.; Baiocchi, G.; Giulini, S.M.; Ansaloni, L.; et al. Peritoneal Carcinomatosis. World J. Gastroenterol. 2013, 19, 6979–6994.
- McMullen, J.R.W.; Selleck, M.; Wall, N.R.; Senthil, M. Peritoneal Carcinomatosis: Limits of Diagnosis and the Case for Liquid Biopsy. Oncotarget 2017, 8, 43481–43490.
- Shariat-Madar, B.; Jayakrishnan, T.T.; Gamblin, T.C.; Turaga, K.K. Surgical Management of Bowel Obstruction in Patients with Peritoneal Carcinomatosis. J. Surg. Oncol. 2014, 110, 666–669.
- Glass, R.L.; LeDuc, R.J. Small Intestinal Obstruction from Peritoneal Carcinomatosis. Am. J. Surg. 1973, 125, 316–317.
- Chu, D.Z.; Lang, N.P.; Thompson, C.; Osteen, P.K.; Westbrook, K.C. Peritoneal Carcinomatosis in Nongynecologic Malignancy. A Prospective Study of Prognostic Factors. Cancer 1989, 63, 364–367.
- Kerscher, A.G.; Chua, T.C.; Gasser, M.; Maeder, U.; Kunzmann, V.; Isbert, C.; Germer, C.T.; Pelz, J.O.W. Impact of Peritoneal Carcinomatosis in the Disease History of Colorectal Cancer Management: A Longitudinal Experience of 2406 Patients over Two Decades. Br. J. Cancer 2013, 108, 1432–1439.
- Yonemura, Y.; Bandou, E.; Kinoshita, K.; Kawamura, T.; Takahashi, S.; Endou, Y.; Sasaki, T. Effective Therapy for Peritoneal Dissemination in Gastric Cancer. Surg. Oncol. Clin. N. Am. 2003, 12, 635–648.
- Thomassen, I.; Bernards, N.; van Gestel, Y.R.; Creemers, G.-J.; Jacobs, E.M.; Lemmens, V.E.; de Hingh, I.H. Chemotherapy as Palliative Treatment for Peritoneal Carcinomatosis of Gastric Origin. Acta. Oncol. 2014, 53, 429–432.
- Sugarbaker, P.H.; Jablonski, K.A. Prognostic Features of 51 Colorectal and 130 Appendiceal Cancer Patients with Peritoneal Carcinomatosis Treated by Cytoreductive Surgery and Intraperitoneal Chemotherapy. Ann. Surg. 1995, 221, 124–132.
- Mahteme, H.; Hansson, J.; Berglund, A.; Påhlman, L.; Glimelius, B.; Nygren, P.; Graf, W. Improved Survival in Patients with Peritoneal Metastases from Colorectal Cancer: A Preliminary Study. Br. J. Cancer 2004, 90, 403–407.
- Verwaal, V.; van Ruth, S.; de Bree, E.; van Sloothen, G.; van Tinteren, H.; Boot, H.; Zoetmulder, F. Randomized Trial of Cytoreduction and Hyperthermic Intraperitoneal Chemotherapy versus Systemic Chemotherapy and Palliative Surgery in Patients with Peritoneal Carcinomatosis of Colorectal Cancer. J. Clin. Oncol. 2003, 21, 3737–3743. Available online: https://pubmed.ncbi.nlm.nih.gov/14551293/ (accessed on 14 July 2022).
- Van Oudheusden, T.R.; Nienhuijs, S.W.; Luyer, M.D.; Nieuwenhuijzen, G.A.; Lemmens, V.E.; Rutten, H.J.; de Hingh, I.H. Incidence and Treatment of Recurrent Disease after Cytoreductive Surgery and Intraperitoneal Chemotherapy for Peritoneally Metastasized Colorectal Cancer: A Systematic Review. Eur. J. Surg. Oncol. 2015, 41, 1269–1277.
- Karunasena, E.; Sham, J.; McMahon, K.W.; Ahuja, N. Genomics of Peritoneal Malignancies. Surg. Oncol. Clin. N. Am. 2018, 27, 463–475.
- Slavin, T.; Neuhausen, S.L.; Rybak, C.; Solomon, I.; Nehoray, B.; Blazer, K.; Niell-Swiller, M.; Adamson, A.W.; Yuan, Y.-C.; Yang, K.; et al. Genetic Gastric Cancer Susceptibility in the International Clinical Cancer Genomics Community Research Network. Cancer Genet. 2017, 216–217, 111–119.
- Ströhlein, M.; Heiss, M.; Jauch, K.-W. The Current Status of Immunotherapy in Peritoneal Carcinomatosis. Expert Rev. Anticancer. Ther. 2016, 16, 1019–1027.
- Kanda, M.; Kodera, Y. Molecular Mechanisms of Peritoneal Dissemination in Gastric Cancer. World J. Gastroenterol. 2016, 22, 6829–6840.
- Li, X.; Ji, Z.; Li, Y. Peritoneal Carcinomatosis Diagnosis and Treatment in China: Focusing on Training and Collaboration. Indian J. Surg. Oncol. 2019, 10, 12–18.
- Sadeghi, B.; Arvieux, C.; Glehen, O.; Beaujard, A.C.; Rivoire, M.; Baulieux, J.; Fontaumard, E.; Brachet, A.; Caillot, J.L.; Faure, J.L.; et al. Peritoneal Carcinomatosis from Non-Gynecologic Malignancies: Results of the EVOCAPE 1 Multicentric Prospective Study. Cancer 2000, 88, 358–363.
- Chia, C.S.; You, B.; Decullier, E.; Vaudoyer, D.; Lorimier, G.; Abboud, K.; Bereder, J.-M.; Arvieux, C.; Boschetti, G.; Glehen, O.; et al. Patients with Peritoneal Carcinomatosis from Gastric Cancer Treated with Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy: Is Cure a Possibility? Ann. Surg. Oncol. 2016, 23, 1971–1979.
- Verwaal, V.J.; Bruin, S.; Boot, H.; van Slooten, G.; van Tinteren, H. 8-Year Follow-up of Randomized Trial: Cytoreduction and Hyperthermic Intraperitoneal Chemotherapy versus Systemic Chemotherapy in Patients with Peritoneal Carcinomatosis of Colorectal Cancer. Ann. Surg. Oncol. 2008, 15, 2426–2432.
- Yao, X.; Ajani, J.; Song, S. Molecular Biology and Immunology of Gastric Cancer Peritoneal Metastasis. Transl. Gastroenterol. Hepatol. 2020, 5, 57.
- Kubicka, U.; Olszewski, W.L.; Tarnowski, W.; Bielecki, K.; Ziółkowska, A.; Wierzbicki, Z. Normal Human Immune Peritoneal Cells: Subpopulations and Functional Characteristics. Scand. J. Immunol. 1996, 44, 157–163.
- Bagheri, V.; Abbaszadegan, M.R.; Memar, B.; Motie, M.R.; Asadi, M.; Mahmoudian, R.A.; Gholamin, M. Induction of T Cell-Mediated Immune Response by Dendritic Cells Pulsed with MRNA of Sphere-Forming Cells Isolated from Patients with Gastric Cancer. Life Sci. 2019, 219, 136–143.
- Fujimori, D.; Kinoshita, J.; Yamaguchi, T.; Nakamura, Y.; Gunjigake, K.; Ohama, T.; Sato, K.; Yamamoto, M.; Tsukamoto, T.; Nomura, S.; et al. Established Fibrous Peritoneal Metastasis in an Immunocompetent Mouse Model Similar to Clinical Immune Microenvironment of Gastric Cancer. BMC Cancer 2020, 20, 1014.
- Park, H.S.; Kwon, W.S.; Park, S.; Jo, E.; Lim, S.J.; Lee, C.-K.; Lee, J.B.; Jung, M.; Kim, H.S.; Beom, S.-H.; et al. Comprehensive Immune Profiling and Immune-Monitoring using Body Fluid of Patients with Metastatic Gastric Cancer. J. Immunother. Cancer 2019, 7, 268.
- Lim, B.; Kim, C.; Kim, J.-H.; Kwon, W.S.; Lee, W.S.; Kim, J.M.; Park, J.Y.; Kim, H.S.; Park, K.H.; Kim, T.S.; et al. Genetic Alterations and their Clinical Implications in Gastric Cancer Peritoneal Carcinomatosis Revealed by Whole-Exome Sequencing of Malignant Ascites. Oncotarget 2016, 7, 8055–8066.
- Sasada, T.; Kimura, M.; Yoshida, Y.; Kanai, M.; Takabayashi, A. CD4+CD25+ Regulatory T Cells in Patients with Gastrointestinal Malignancies: Possible Involvement of Regulatory T Cells in Disease Progression. Cancer 2003, 98, 1089–1099.
- Wada, J.; Suzuki, H.; Fuchino, R.; Yamasaki, A.; Nagai, S.; Yanai, K.; Koga, K.; Nakamura, M.; Tanaka, M.; Morisaki, T.; et al. The Contribution of Vascular Endothelial Growth Factor to the Induction of Regulatory T-Cells in Malignant Effusions. Antcancer Res. 2009, 29, 881–888.
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355.
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723.
- Chia, D.K.A.; Gwee, Y.X.; Sundar, R. Resistance to Systemic Immune Checkpoint Inhibition in the Peritoneal Niche. J. Immunother. Cancer 2022, 10, e004749.
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.-Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 2017, PO.17.00073.
- Casak, S.J.; Marcus, L.; Fashoyin-Aje, L.; Mushti, S.L.; Cheng, J.; Shen, Y.-L.; Pierce, W.F.; Her, L.; Goldberg, K.B.; Theoret, M.R.; et al. FDA Approval Summary: Pembrolizumab for the First-Line Treatment of Patients with MSI-H/DMMR Advanced Unresectable or Metastatic Colorectal Carcinoma. Clin. Cancer Res. 2021, 27, 4680–4684.
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10.
- Kumagai, Y.; Futoh, Y.; Miyato, H.; Ohzawa, H.; Yamaguchi, H.; Saito, S.; Kurashina, K.; Hosoya, Y.; Lefor, A.K.; Sata, N.; et al. Effect of Systemic or Intraperitoneal Administration of Anti-PD-1 Antibody for Peritoneal Metastases from Gastric Cancer. Vivo 2022, 36, 1126–1135.
- Ma, Z.; Li, W.; Yoshiya, S.; Xu, Y.; Hata, M.; El-Darawish, Y.; Markova, T.; Yamanishi, K.; Yamanishi, H.; Tahara, H.; et al. Augmentation of Immune Checkpoint Cancer Immunotherapy with IL18. Clin. Cancer Res. 2016, 22, 2969–2980.
- Lee, Y.S.; Lee, W.S.; Kim, C.W.; Lee, S.J.; Yang, H.; Kong, S.J.; Ning, J.; Yang, K.-M.; Kang, B.; Kim, W.R.; et al. Oncolytic Vaccinia Virus Reinvigorates Peritoneal Immunity and Cooperates with Immune Checkpoint Inhibitor to Suppress Peritoneal Carcinomatosis in Colon Cancer. J. Immunother. Cancer 2020, 8, e000857.
- Wei, H.; Zhao, L.; Li, W.; Fan, K.; Qian, W.; Hou, S.; Wang, H.; Dai, M.; Hellstrom, I.; Hellstrom, K.E.; et al. Combinatorial PD-1 Blockade and CD137 Activation Has Therapeutic Efficacy in Murine Cancer Models and Synergizes with Cisplatin. PLoS ONE 2013, 8, e84927.
- Guo, Z.; Wang, X.; Cheng, D.; Xia, Z.; Luan, M.; Zhang, S. PD-1 Blockade and OX40 Triggering Synergistically Protects against Tumor Growth in a Murine Model of Ovarian Cancer. PLoS ONE 2014, 9, e89350.
- Lu, L.; Xu, X.; Zhang, B.; Zhang, R.; Ji, H.; Wang, X. Combined PD-1 Blockade and GITR Triggering Induce a Potent Antitumor Immunity in Murine Cancer Models and Synergizes with Chemotherapeutic Drugs. J. Transl. Med. 2014, 12, 36.
- Yamaguchi, T.; Fushida, S.; Yamamoto, Y.; Tsukada, T.; Kinoshita, J.; Oyama, K.; Miyashita, T.; Tajima, H.; Ninomiya, I.; Munesue, S.; et al. Tumor-Associated Macrophages of the M2 Phenotype Contribute to Progression in Gastric Cancer with Peritoneal Dissemination. Gastric Cancer 2016, 19, 1052–1065.
- Nakamura, Y.; Kinoshita, J.; Yamaguchi, T.; Aoki, T.; Saito, H.; Hamabe-Horiike, T.; Harada, S.; Nomura, S.; Inaki, N.; Fushida, S. Crosstalk between Cancer-Associated Fibroblasts and Immune Cells in Peritoneal Metastasis: Inhibition in the Migration of M2 Macrophages and Mast Cells by Tranilast. Gastric Cancer 2022, 25, 515–526.
- Bellone, S.; Siegel, E.R.; Cocco, E.; Cargnelutti, M.; Silasi, D.-A.; Azodi, M.; Schwartz, P.E.; Rutherford, T.J.; Pecorelli, S.; Santin, A.D. Overexpression of Epithelial Cell Adhesion Molecule in Primary, Metastatic, and Recurrent/Chemotherapy-Resistant Epithelial Ovarian Cancer: Implications for Epithelial Cell Adhesion Molecule-Specific Immunotherapy. Int. J. Gynecol. Cancer 2009, 19, 860–866.
- Köbel, M.; Kalloger, S.E.; Boyd, N.; McKinney, S.; Mehl, E.; Palmer, C.; Leung, S.; Bowen, N.J.; Ionescu, D.N.; Rajput, A.; et al. Ovarian Carcinoma Subtypes are Different Diseases: Implications for Biomarker Studies. PLoS Med. 2008, 5, e232.
- Andersson, Y.; Engebraaten, O.; Fodstad, Ø. Synergistic Anti-Cancer Effects of Immunotoxin and Cyclosporin in Vitro and in Vivo. Br. J. Cancer 2009, 101, 1307–1315.
- Andersson, Y.; Juell, S.; Fodstad, Ø. Downregulation of the Antiapoptotic MCL-1 Protein and Apoptosis in MA-11 Breast Cancer Cells Induced by an Anti-Epidermal Growth Factor Receptor-Pseudomonas Exotoxin a Immunotoxin. Int. J. Cancer 2004, 112, 475–483.
- Andersson, Y.; Engebraaten, O.; Juell, S.; Aamdal, S.; Brunsvig, P.; Fodstad, Ø.; Dueland, S. Phase I Trial of EpCAM-Targeting Immunotoxin MOC31PE, Alone and in Combination with Cyclosporin. Br. J. Cancer 2015, 113, 1548–1555.
- Frøysnes, I.S.; Andersson, Y.; Larsen, S.G.; Davidson, B.; Øien, J.-M.T.; Olsen, K.H.; Giercksky, K.-E.; Julsrud, L.; Fodstad, Ø.; Dueland, S.; et al. Novel Treatment with Intraperitoneal MOC31PE Immunotoxin in Colorectal Peritoneal Metastasis: Results From the ImmunoPeCa Phase 1 Trial. Ann. Surg. Oncol. 2017, 24, 1916–1922.
- Wiiger, M.T.; Bideli, H.; Fodstad, O.; Flatmark, K.; Andersson, Y. The MOC31PE Immunotoxin Reduces Cell Migration and Induces Gene Expression and Cell Death in Ovarian Cancer Cells. J. Ovarian Res. 2014, 7, 23.
- Flatmark, K.; Guldvik, I.J.; Svensson, H.; Fleten, K.G.; Flørenes, V.A.; Reed, W.; Giercksky, K.-E.; Fodstad, Ø.; Andersson, Y. Immunotoxin Targeting EpCAM Effectively Inhibits Peritoneal Tumor Growth in Experimental Models of Mucinous Peritoneal Surface Malignancies. Int. J. Cancer 2013, 133, 1497–1506.
- Ströhlein, M.A.; Heiss, M.M. Intraperitoneal Immunotherapy to Prevent Peritoneal Carcinomatosis in Patients with Advanced Gastrointestinal Malignancies. J. Surg. Oncol. 2009, 100, 329–330.
- Jäger, M.; Schoberth, A.; Ruf, P.; Hess, J.; Hennig, M.; Schmalfeldt, B.; Wimberger, P.; Ströhlein, M.; Theissen, B.; Heiss, M.M.; et al. Immunomonitoring Results of a Phase II/III Study of Malignant Ascites Patients Treated with the Trifunctional Antibody Catumaxomab (Anti-EpCAM × Anti-CD3). Cancer Res. 2012, 72, 24–32.
- Ströhlein, M.A.; Heiss, M.M. Immunotherapy of Peritoneal Carcinomatosis. Cancer Treat. Res. 2007, 134, 483–491.
- Chelius, D.; Ruf, P.; Gruber, P.; Plöscher, M.; Liedtke, R.; Gansberger, E.; Hess, J.; Wasiliu, M.; Lindhofer, H. Structural and Functional Characterization of the Trifunctional Antibody Catumaxomab. MAbs 2010, 2, 309–319.
- Seimetz, D. Novel Monoclonal Antibodies for Cancer Treatment: The Trifunctional Antibody Catumaxomab (Removab®). J. Cancer 2011, 2, 309–316.
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The Trifunctional Antibody Catumaxomab for the Treatment of Malignant Ascites Due to Epithelial Cancer: Results of a Prospective Randomized Phase II/III Trial. Int. J. Cancer 2010, 127, 2209–2221.
- Heiss, M.M.; Ströhlein, M.A.; Jäger, M.; Kimmig, R.; Burges, A.; Schoberth, A.; Jauch, K.-W.; Schildberg, F.-W.; Lindhofer, H. Immunotherapy of Malignant Ascites with Trifunctional Antibodies. Int. J. Cancer 2005, 117, 435–443.
- Burges, A.; Wimberger, P.; Kümper, C.; Gorbounova, V.; Sommer, H.; Schmalfeldt, B.; Pfisterer, J.; Lichinitser, M.; Makhson, A.; Moiseyenko, V.; et al. Effective Relief of Malignant Ascites in Patients with Advanced Ovarian Cancer by a Trifunctional Anti-EpCAM × Anti-CD3 Antibody: A Phase I/II Study. Clin. Cancer Res. 2007, 13, 3899–3905.
- Mackey, J.R.; Venner, P.M. Malignant Ascites: Demographics, Therapeutic Efficacy and Predictors of Survival. Can. J. Oncol. 1996, 6, 474–480.
- Wimberger, P.; Gilet, H.; Gonschior, A.-K.; Heiss, M.M.; Moehler, M.; Oskay-Oezcelik, G.; Al-Batran, S.-E.; Schmalfeldt, B.; Schmittel, A.; Schulze, E.; et al. Deterioration in Quality of Life (QoL) in Patients with Malignant Ascites: Results from a Phase II/III Study Comparing Paracentesis plus Catumaxomab with Paracentesis Alone. Ann. Oncol. 2012, 23, 1979–1985.
- Hollingsworth, R.E.; Jansen, K. Turning the Corner on Therapeutic Cancer Vaccines. NPJ Vaccines 2019, 4, 7.
- Le, D.T.; Pardoll, D.M.; Jaffee, E.M. Cellular Vaccine Approaches. Cancer J. 2010, 16, 304–310.
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing Oncolytic Virotherapy in Cancer Treatment. Nat. Rev. Drug. Discov. 2019, 18, 689–706.
- Osipov, A.; Murphy, A.; Zheng, L. From Immune Checkpoints to Vaccines: The Past, Present and Future of Cancer Immunotherapy. Adv. Cancer Res. 2019, 143, 63–144.
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292.
- Slingluff, C.L. The Present and Future of Peptide Vaccines for Cancer: Single or Multiple, Long or Short, Alone or in Combination? Cancer J. 2011, 17, 343–350.
- Liu, J.; Miao, L.; Sui, J.; Hao, Y.; Huang, G. Nanoparticle Cancer Vaccines: Design Considerations and Recent Advances. Asian J. Pharm. Sci. 2020, 15, 576–590.
- Ai, Y.-Q.; Cai, K.; Hu, J.-H.; Jiang, L.-W.; Gao, Y.-R.; Zhao, H.; Jia, S.-C. The Clinical Effects of Dendritic Cell Vaccines Combined with Cytokine-Induced Killer Cells Intraperitoneal Injected on Patients with Malignant Ascites. Int. J. Clin. Exp. Med. 2014, 7, 4272–4281.
- Geller, M.A.; Knorr, D.A.; Hermanson, D.A.; Pribyl, L.; Bendzick, L.; McCullar, V.; Miller, J.S.; Kaufman, D.S. Intraperitoneal Delivery of Human Natural Killer Cells for Treatment of Ovarian Cancer in a Mouse Xenograft Model. Cytotherapy 2013, 15, 1297–1306.
- Dobrzanski, M.J.; Rewers-Felkins, K.A.; Quinlin, I.S.; Samad, K.A.; Phillips, C.A.; Robinson, W.; Dobrzanski, D.J.; Wright, S.E. Autologous MUC1-Specific Th1 Effector Cell Immunotherapy Induces Differential Levels of Systemic TReg Cell Subpopulations That Result in Increased Ovarian Cancer Patient Survival. Clin. Immunol. 2009, 133, 333–352.
- Deng, J.; Wang, L.; Chen, H.; Li, L.; Ma, Y.; Ni, J.; Li, Y. The Role of Tumour-Associated MUC1 in Epithelial Ovarian Cancer Metastasis and Progression. Cancer Metastasis Rev. 2013, 32, 535–551.
- Alkayyal, A.A.; Tai, L.-H.; Kennedy, M.A.; de Souza, C.T.; Zhang, J.; Lefebvre, C.; Sahi, S.; Ananth, A.A.; Mahmoud, A.B.; Makrigiannis, A.P.; et al. NK-Cell Recruitment Is Necessary for Eradication of Peritoneal Carcinomatosis with an IL12-Expressing Maraba Virus Cellular Vaccine. Cancer Immunol. Res. 2017, 5, 211–221.
- Gross, G.; Waks, T.; Eshhar, Z. Expression of Immunoglobulin-T-Cell Receptor Chimeric Molecules as Functional Receptors with Antibody-Type Specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028.
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152.
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398.
- Dai, H.; Wang, Y.; Lu, X.; Han, W. Chimeric Antigen Receptors Modified T-Cells for Cancer Therapy. J. Natl. Cancer Inst. 2016, 108, djv439.
- Kershaw, M.H.; Westwood, J.A.; Darcy, P.K. Gene-Engineered T Cells for Cancer Therapy. Nat. Rev. Cancer 2013, 13, 525–541.
- Chmielewski, M.; Hombach, A.A.; Abken, H. Antigen-Specific T-Cell Activation Independently of the MHC: Chimeric Antigen Receptor-Redirected T Cells. Front. Immunol. 2013, 4, 371.
- Katz, S.C.; Point, G.R.; Cunetta, M.; Thorn, M.; Guha, P.; Espat, N.J.; Boutros, C.; Hanna, N.; Junghans, R.P. Regional CAR-T Cell Infusions for Peritoneal Carcinomatosis Are Superior to Systemic Delivery. Cancer Gene Ther. 2016, 23, 142–148.
- Ang, W.X.; Li, Z.; Chi, Z.; Du, S.-H.; Chen, C.; Tay, J.C.K.; Toh, H.C.; Connolly, J.E.; Xu, X.H.; Wang, S. Intraperitoneal Immunotherapy with T Cells Stably and Transiently Expressing Anti-EpCAM CAR in Xenograft Models of Peritoneal Carcinomatosis. Oncotarget 2017, 8, 13545–13559.
- García-Olmo, D.; Villarejo Campos, P.; Barambio, J.; Gomez-Heras, S.G.; Vega-Clemente, L.; Olmedillas-Lopez, S.; Guadalajara, H.; Garcia-Arranz, M. Intraperitoneal Collagenase as a Novel Therapeutic Approach in an Experimental Model of Colorectal Peritoneal Carcinomatosis. Sci. Rep. 2021, 11, 503.
- Koneru, M.; Purdon, T.; Spriggs, D.; Koneru, S.; Brentjens, R. IL-12 Secreting Tumor-Targeted Chimeric Antigen Receptor T Cells Eradicate Ovarian Tumors in Vivo. Oncoimmunology 2015, 4, e994446.