Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, Research & Experimental
The p53 tumor suppressor molecule is a critical regulator of cell signaling pathways, cellular stress responses, DNA repair, and apoptosis. However, viruses can activate or inhibit p53 during viral infections to enhance viral replication and spread. Given its pivotal role in cell physiology, p53 represents a potential target for anti-coronavirus drugs.
- p53
- coronavirus
- antiviral
1. Introduction
Coronaviruses (CoVs) are a diverse family of single-stranded positive-sense enveloped RNA viruses that can induce a range of issues, including the stress response of host cells, cell cycle changes, cell apoptosis, and host immune dysregulation and inflammation [1]. They can infect humans and various vertebrates, leading to severe public health problems and economic losses worldwide [2,3]. Coronaviruses were first isolated from chickens in 1937. Usually, the host range of CoVs is very narrow, and there are six coronaviruses other than the 2019 coronavirus disease (COVID-19) that can infect humans [4]. However, the multiple coronavirus pandemics that have occurred in recent years, such as the 2019 SARS-CoV-2, the 2012 middle east respiratory syndrome (MERS), and the 2002 severe acute respiratory syndrome (SARS) outbreaks, have demonstrated the potential for the zoonotic and human-to-human transmission of emerging coronaviruses [5,6].
The p53 tumor suppressor is a crucial molecule attending to the regulation of various basic cell signaling, including cell cycle, DNA repair, apoptosis, and cellular stress response [10,11,12]. Moreover, apart from its role in regulating cell growth and apoptosis, p53 also plays a crucial role in various cellular processes, including metabolism, autophagy, and innate immunity [13]. This protein can directly or indirectly influence cellular metabolism, impacting the production and consumption of energy within the cell [14]. Additionally, p53 is involved in the process of autophagy, which plays a key role in removing damaged cellular components and recycling cellular building blocks [15] (Figure 1).
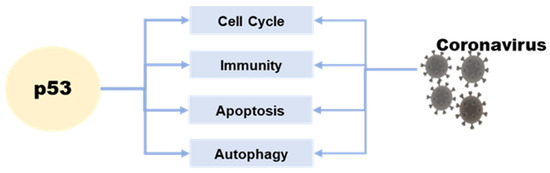
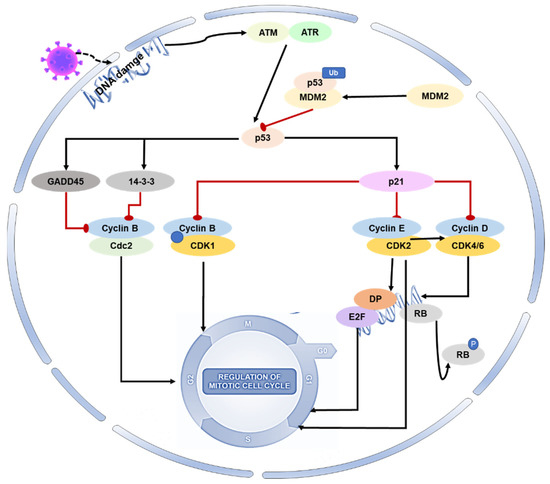
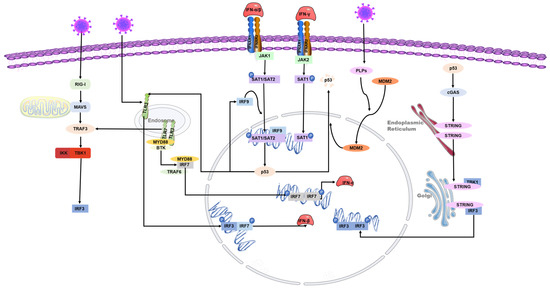
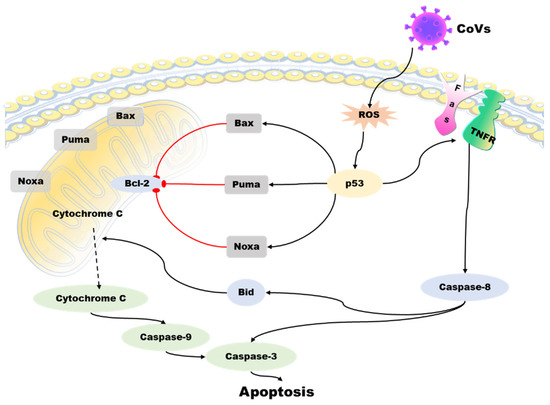
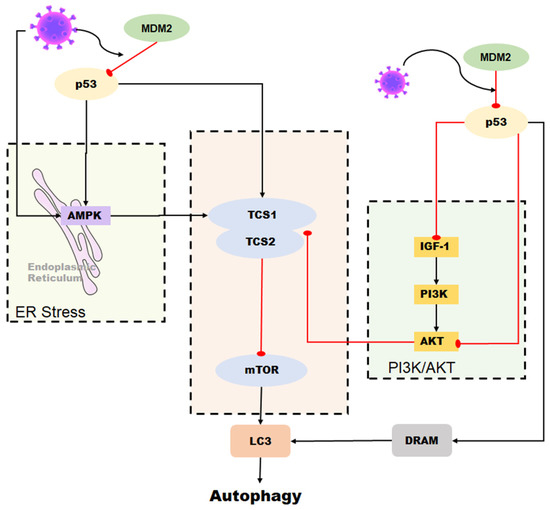
Figure 1. The relationship between p53 and coronavirus.
2. p53 Affects Coronaviruses by Regulating the Cell Cycle
Cell cycle progression may be an advanced and orderly regulated method. Throughout this method, multiple units work alongside to make sure of smart division and proliferation [26]. Cyclin-dependent kinases (CDK) and cyclins mediate the cell cycle progression [27]. Inhibition or absence of CDK activity can result in cells’ arrest in the G1 phase and their entry into a quiescent state [28]. The G1-S and G2-M phases of the cell cycle provide protection against both exogenous and endogenous agents that can cause DNA damage [29]. The molecular level changes in these two stages are complex and susceptible to environmental conditions caused by coronaviruses [30]. P21 is an inhibitor of CDKs and regulates the cell cycle by interfering with cyclins [31]. The transition from the S phase to the G1 phase is facilitated by cyclins. CDKs are inhibited by p21WAF1/CIP1, which causes low phosphorylation of retinoblastoma (Rb) and prevents E2F release, thereby blocking the transformation of the G1-S and G2-M phases. G2-M arrest is mainly regulated by the p53-p21-DREAM pathway, which indirectly inhibits the transcription of cell cycle genes such as CCNB2, KIF23, and PLK4 [32].
Regulation of the host cell cycle could be a common strategy several viruses use for infectious agent replication (creating an excellent cellular environment). Coronavirus infection not only induces oxidative stress and DNA damage in host cells, but it can also interfere with the host cell cycle by directly altering the activity of p53 or its upstream and downstream proteins [33,34,35,36,37]. SARS-CoV-2 N protein can induce acute kidney injury by arresting the cell’s G1 cycle [38]. With the deepening of coronavirus–host interaction research, the p53-DREAM pathway provides a novel antiviral strategy. Porcine epidemic diarrhea virus (PEDV) N protein induces S-phase arrest in host cells by activating p53 and binding downstream protein DREAM to CHR [39]. Infection with PEDV can modify the expression of proteins involved in cell cycle regulation, such as p21, CDK1, CDK2, CDK4, cyclin A, and cyclin E. The viral infection can cause cell cycle arrest in the G0-G1 phase, which can be restored by inhibiting the p53 signaling pathway. This leads to the downregulation of p21 and associated cyclin/CDK proteins [43] (Figure 2).
Figure 2. Role of p53 on the cell cycle during coronavirus infection. During viral infection, p53 is activated and initiates a cascade of events that result in cell cycle arrest. Specifically, activated p53 mediates the protein p21 to form complexes with CDKs, resulting in the inhibition of cyclin–CDK activity. This also leads to a reduction in Rb phosphorylation, thereby maintaining the stability of the RB–E2F complex and inhibiting the transcription of cell cycle genes. Additionally, p53 mediates the proteins 14-3-3 and GADD45 to inhibit cyclin–CDK1 activity, resulting in cell arrest in the G2-M phase.
3. The Interaction between p53 and Interferon
IFNs possess diverse biological activities, which include antiviral effects, antiproliferative effects, and the activation of immune cell cytotoxicity. IFNs are central to antiviral immunity and can inhibit coronaviruses’ replication. Cells produce type I IFN (primarily IFN-α and IFN-β) in response to viral infections, which is critical for immunity against many types of viruses. The transcription of antiviral-related genes is induced by IFN-I, which can be triggered by viral recognition sensors such as toll-like receptors and RNA helicases such as the RNA helicase retinoic acid-inducible gene I (RIG-I). The activation of interferon regulatory factor (IRF) leads to the production of type I interferon [44]. IFNAR1 and IFNAR2 form a type I IFN receptor that recognizes and binds to type I IFN, giving bystander cells antiviral effects.
p53 also contributes to the increased release of IFN-1 from virus-infected cells [47]. IRF9 was confirmed to be a p53 target gene, suggesting that type I IFN can not only increase the expression of p53 by activating IRF9 but also that p53 can activate IRF9. IRF9 continues to activate retinoic acid inducer 1 (RIG-I) and ISRE-dependent genes such as IRF7 [47,48]. IRF3 and IRF7 play a crucial role in inducing the expression of type I interferon genes downstream of pattern recognition receptors. These transcription factors bind to the promoters of IFN-α and IFN-β through homologous or heterologous interactions, thereby controlling their expression [49].
p53 can inhibit the replication of coronaviruses and shows antiviral activity in vivo; this effect may be due to its ability to activate natural immune pathways. Previous studies have shown that several coronaviruses, such as SARS-CoV-2, SAR-CoV, and human coronavirus NL63 infections, can only induce deficient levels of IFN-I [51,52,53], which is likely to lead to uninhibited viral replication and damage to the immune system. Low-level IFN responses may be a means by which coronaviruses evade immunity. The lack of adequate IFN response may be due to the decrease of p53 hydrolyzed by the coronavirus papain-like proteases (PLPs). PLPs are a class of cysteine proteases that inhibit innate immunity by stabilizing the binding of MDM2 and p53 to cause the ubiquitination of p53 [54]. The SARS-unique domain (SUD) and papain-like proteases (PLPs) interact with cell E3 ubiquitin ligase and CHY zinc-finger domain-containing 1 (RCHY1) to facilitate their activities. SUD and PLPs target p53 with E3 ubiquitin ligase RCHY1 to degrade p53. The degradation of p53 decreases the level of earthly IFN [55]. In other words, coronaviruses can evade the host’s natural immune defenses by degrading p53 through their own proteins. At the same time, the application of small molecule inhibitors of MDM2, such as nutlin-3 and idasanutlin, can promote the stable presence of p53 in cells, help regulate the IFN signaling pathway, and inhibit the replication of coronaviruses [56] (Figure 3).
Figure 3. Role of p53 in the regulation of interferon during coronavirus infection. The toll-like receptor and RNA helicase retinoic acid-inducing gene I (RIG-I) are activated after the RNA virus invasion of cells. IRF3 is phosphorylated to produce type I interferon. The JAK-STAT signaling pathway is subsequently activated, and the transcription factors STAT-1, STAT-2, and IFN regulatory factor 9 (IRF9) bind to induce p53 expression. IRF9 is also a p53 target gene and is regulated by p53. Virus infection activates p53-mediated TLR3, phosphorylates downstream IRF3 and IRF7 to form dimers, and promotes the synthesis of type I interferon. p53 also activates IRF3 through the p53-cgas-STING pathway. In addition, coronaviruses can evade the host’s natural immune defenses by degrading p53 through their papain-like proteases (PLPs).
4. p53 Affects Coronavirus-Associated Apoptosis
Apoptosis is a programmed death mechanism evolved by cells in response to the complex external environment and self-injury. This mechanism is dangerous but effective for living organisms. Due to the particularity and importance of apoptosis, there is a complicated regulatory mechanism network, and p53 is one of the links. Apoptosis is primarily controlled by intrinsic and extrinsic pathways. The intrinsic pathway of apoptosis involves a complex downstream signaling network that is regulated by p53 [60]. The TNFR family, including the death receptor and Fas, initiates the extrinsic pathway of apoptosis by activating the formation of the death-inducing signaling complex (DISC). This leads to the activation of caspases, including caspase-8 and caspase-3, ultimately resulting in apoptosis [61]. The Bcl-2 family of proteins regulates the intrinsic apoptotic pathway, and it includes anti-apoptotic and pro-apoptotic members. Anti-apoptotic proteins, such as Bcl-XL, can perform anti-apoptotic functions due to their structural similarity to Bcl-2. In contrast, pro-apoptotic proteins such as Bax and Bak can antagonize the anti-apoptotic function of Bcl-2 because they share a similar structure with Bcl-2 and Bcl-XL. The external apoptotic pathway involves the activation of caspases, including caspase-8 and caspase-3, through the death-inducing signaling complex (DISC) formation initiated by the tumor necrosis factor receptor (TNFR) family (such as death receptor and Fas), leading to apoptosis. P53 plays a crucial role in regulating the intrinsic pathway of apoptosis [62]. Interestingly, Bax, Noxa, PUMA (p53-up-regulated apoptosis regulator), and BH3 interaction domain death agonists (BID) are important targets for p53. p53 can upregulate the expression level of these proteins, thus inducing apoptosis [63]. Other studies have shown that p53 can directly stimulate mitochondria to release high ROS and cause cell apoptosis [64] (Figure 4).
Figure 4. Role of p53 in the regulation of apoptosis during coronavirus infection. The virus induces ROS accumulation in cells and activates p53 protein, which activates the pro-apoptotic proteins of the Bcl-2 family -Bax, Noxa, PUMA, and BID, antagonizing the anti-apoptotic effect of BCL-2. Then, cytochrome C from mitochondria spills over, triggering a cascade of caspase proteins, leading to apoptosis. p53 can also act on the tumor necrosis factor receptor (TNFR) family to activate caspase-3 and caspase-8 through the exogenous apoptotic pathway and cause apoptosis.
5. p53 Affects Coronavirus-Associated Autophagy
Autophagy is a process by which cells recycle cellular components and eliminate damaged organelles or misfolded proteins. It is also a critical mechanism for the innate immune response against viral infection. p53 regulates autophagy through transcription-dependent and transcription-independent mechanisms. It can stimulate autophagy by upregulating genes such as damage-regulated autophagy modulator (DRAM) and sestrin2. Additionally, p53 can interact directly with autophagy-related proteins, such as LC3 [70,71].
In conclusion, the relationship between p53 and autophagy during coronavirus infection is complex and context-dependent. While p53 can promote autophagy as a host defense mechanism against viral infection, some coronaviruses can also manipulate the p53 pathway to inhibit autophagy and promote their own replication (Figure 5). Further research is necessary to fully comprehend the interplay between p53 and autophagy during coronavirus infection and identify potential targets for therapeutic intervention.
Figure 5. Role of p53 in the regulation of autophagy during coronavirus infection. Autophagy can be induced by p53 through the DRAM protein. In some coronaviruses, autophagy is activated by p53-mediated DRAM protein as well as through the AMPK pathway and the PI3K/AKT pathway. However, some coronaviruses can reduce the stability of p53 in cells and inhibit p53-mediated autophagy by upregulating the expression of MDM2.
6. Coronavirus Proteins Interact with p53
Through long evolution, coronaviruses induce cell cycle arrest, apoptosis, autophagy in the invasion into host cells, genome replication, the assembly and release of virions, and immune escape [77,78,79,80]. Four major proteins, nucleocapsid protein (N), membrane protein (M), envelope protein (E), and spike protein (S), make up coronavirus particles and are called structural proteins. [81]. These proteins help coronaviruses complete their viral cycle. The coronavirus N protein regulates viral replication and transcription and plays a vital role in the viral integration of genomic RNA into progeny virions [82]. The M and E proteins and membranes from host organelles form the virion envelope. The S protein is embedded in the envelope. The function of the coronavirus highly glycosylated S protein is to help the virus adhere, enter, fuse the virus envelope to the cell membrane, and neutralize antibodies [83].
While these proteins play their functions, p53 also becomes an important participant. Upregulation of p53 expression induced by transmissible gastroenteritis virus (TGEV) nucleocapsid protein leads to cell cycle arrest and apoptosis. Additionally, TGEV N protein induces changes in the expression of CDK1, CDK2, and cyclin B2, as well as cytochrome c translocation and Bax activation. However, inhibition of p53 can reverse these expression changes and weaken the TGEV N-protein-induced cell cycle arrest and apoptosis [85].
7. p53 Is a Potential Target for Anti-Coronavirus Drugs
Effective anti-coronavirus drugs rely on identifying potential targets for antiviral drugs, which are often aimed at inhibiting key viral replication proteins [88,89]. Nevertheless, RNA virus replication usually has a high error rate (or low viral fidelity), leading to viruses existing in distinct populations of genomic mutants or “quasispecies”, which means viruses can quickly develop resistance to drugs or vaccines [90].
In recent years, more and more researchers have focused on the host cell pathway and virus–host interaction [94,95,96]. Tp53 is a crucial tumor suppressor gene in host cells. Its gene and p53 protein, encoded by Tp53, are widespread antitumor drug targets. p53-targeting therapies include but are not limited to compounds that restore/reactivate wild-type p53 function or eliminate mutant p53 [97,98,99,100]. However, p53 is not a common target in antiviral drug development. With the deepening of research on p53 in antitumor drugs, researchers have also observed its potential for antiviral effect [54,101,102]. During viral infection, p53 is activated by post-translational modifications (such as phosphorylation, acetylation, prime acylation, and methylation) with the participation of many cofactors. Activated p53 triggers a cascade that regulates the transcription and expression of its downstream targets. p53 mediates downstream targets to regulate cell cycle arrest, metabolism, apoptosis, iron death, autophagy, stem cell differentiation, DNA repair, and senescence [47,103,104,105].
This entry is adapted from the peer-reviewed paper 10.3390/ijms24076371
This entry is offline, you can click here to edit this entry!