FokI–dCas9 (fdCas9) or RNA-guided FokI nuclease (RFN) is a genome editing tool engineered by fusing the catalytic domain of the FokI endonuclease to the N-terminal catalytic inactive Cas9 protein. The fdCas9 system is active only as a heterodimer, requiring the simultaneous binding of two fdCas9/sgRNAs monomers at adjacent target sites in a PAM-out orientation, and a specific spacer distance separating the two binding sites of the two sgRNAs. The five engineered and published constructs induce significant gene editing activities and specificities, as tested on various gene targets and overcome the off-target effects associated with other genome editing tools, including ZFNs, TALENs, and CRISPR/Cas systems.
- dead Cas9
- FokI endonuclease
- FokI-dCas9
- dual guide RNA
- fdCas9 variants
1. Introductions
The CRISPR/Cas9 system was first identified as a part of the bacterial immune response against bacteriophages and is now repurposed as a powerful genome-engineering tool that has been successfully used across many eukaryotic species[1][2][3][4][5][6]. Generally, the system is composed of an endonuclease Cas protein that drives DNA cleavage activity and a single guide RNA molecule (sgRNA) that determines the system’s specificity through binding to the targeted gene, following the Watson–Crick base-pairing rule[2][3][6]. A variety of Cas proteins have been identified, each recognizing different protospacer adjacent motif (PAM) sequences at the target sequence [7]. In the case of the CRISPR/Cas9 system, the Streptococcus pyogenes Cas9 (SpCas9) endonuclease recognizes the 5′-NGG-3′ PAM sequence, where N could be any nucleotide[8]. DSBs are created through the two HNH and RuvC endonuclease domains of SpCas9, cleaving the complementary strand and the noncomplementary strand, respectively (Figure 1)[8] . The sgRNA required for the activation of the CRISPR/Cas9 system is composed of a 17–20-nucleotide CRISPR RNA (crRNA) targeting sequence that is a complementary target site and the 85-nucleotide transactivating CRISPR RNA (tracrRNA) processing sequence, which provides structural stability of the ribonucleoprotein complex (RNPC)[9][10]. Upon PAM recognition, sgRNA binding, and the formation of the RNPC, the two catalytic nuclease domains induce DSB 3 to 4 nucleotides upstream of the PAM site, resulting in the activation of DNA repair machinery[11][12][13].
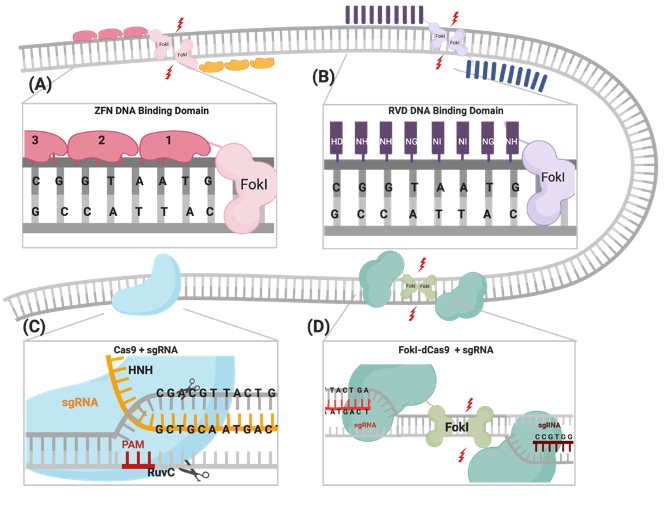
Figure 1. Illustrations of different genome engineering tools. (A) Zinc fingers nucleases (ZFNs) bind to the target sequence through 3-4 zinc finger proteins, and (B) transcription activator-like effector nucleases (TALENs) through repeat variable diresidues (RVDs), both of which inducing double-stranded breaks (DSBs) through heterodimerization and the catalytic activity of the fused FokI. (C) CRISPR–Cas9 binds to the DNA target through PAM sequences and the complementary sgRNA site, where the catalytic activity is mediated through HNH and RuvC domains. (D) FokI–dCas9 (inactive “dead” form of Cas9) binds to the target site through two sgRNAs that are positioned in a PAM-out orientation, and the catalytic activity is mediated through the dimerization of fused FokI endonuclease.
One of the most important advantages of the CRISPR/Cas9 system is the robustness and ease of programming the system through redesigning the sgRNA, the crRNA in particular, into new genes of interest, with the protein constructs remaining constant. In ZFNs and TALENs, complex molecular cloning methods and substantial protein engineering are required with every target design[14]. Although enhanced versions of ZFNs and TALENs have slightly improved their multiplexing efficiencies, the CRISPR system is the easiest tool to multiplex and generate simultaneous gene edits[1][15][16][17][18]. Nevertheless, there are some limitations of using the CRISPR/Cas9 system in genome engineering, which include PAM sequence availability at the target sites and, most importantly, the increased frequency of nonspecific off-target mutations reported in many studies, compared to ZFNs and TALENs (Table 1)[19][20][21][22][23].
Table 1. Comparison between different gene-editing tools.
|
Zinc Finger Nucleases (ZFNs) |
Transcription Activator-Like Effector Nucleases (TALENs) |
CRISPR/Cas Systems |
||
|
CRISPR Associated Endonuclease (Cas9) |
FokI Dead Cas9 Endonuclease (FokI–dCas9) |
|||
|
DNA catalytic domain |
FokI |
FokI |
RuvC and HNH |
FokI |
|
DNA recognition |
DNA: Protein |
DNA: RNA |
||
|
unit of target recognition |
Pairs of ZFNs (via the ZF motifs) |
Pairs of TALENs (via RVD tandem repeat). |
One 17–20 bp sgRNA |
Pairs of 19–20 bp sgRNAs |
|
Recognized target size |
Recognizes 18-24 bp |
Recognizes 30–40 bp. |
Recognizes NGG PAM sequence + 17–20 bp |
Recognizes two NGG PAM sequences + 38–40 bp |
|
Specificity |
Tolerates few positional mismatches |
Tolerates both positional and multiple consecutive mismatches |
Enhanced specificity due to the dual sgRNAs requirement of |
|
|
Spacer size |
5–7 bp |
14–16 bp |
No spacer required |
13–18 bp and/or 26 bp |
|
Ease of delivery |
Limited delivery due to the difficulty of linking ZF modules |
Difficult delivery due to cDNA size and extensive TALEs repeats |
Easily delivered using standard delivery and cloning techniques |
Harder to deliver due to increased size of construct and added components |
|
Limitation |
Off-target effects Limited delivery due to size constraints |
Off-target effects Expensive |
Off-target effects due to mismatch tolerance PAM sequence availability |
Difficult to deliver A strict system with many obligatory requirements |
|
Multiplexing |
Difficult |
Easy, can form multiplexes directed to multiple genes |
||
1.1. Cas9 Variants
Cas9 Nickases
SpCas9 nickases are partially inactive, with either HNH or RuvC endonuclease activity being abolished by the variants D10A and H840A, respectively[24]. These nickases can produce DNA single-strand nicks instead of DNA DSBs, which correlates to the decreased off-target mutation rates when compared to wild-type (WT) Cas9[25]. Genome engineering mediated by Cas9 nickases is inherently more complicated than a WT Cas9[25][26][27]. The requirements are for two sgRNAs functioning simultaneously to generate DSBs, with the predicted overhang lengths and polarities, and PAMs in an outward orientation[26][27]. It is noteworthy that Cas9 D10A-mediated genome engineering is more robust when the two cleavage sites are 37 to 68 bp apart, while Cas9 H840A favors a distance of 51 to 68 bp[28]. Although Cas9 nickases have shown a great decrease in known off-target genes, deep sequencing shows elevated levels of point mutations at the target site, indicating that DNA nicks are still considered mutagenic even when nickases/sgRNA complexes are expressed monomerically[22][26][29][30][31].
Inactive Cas9 (Dead Cas9)
Another form of spCas9 that has been introduced is the inactive dCas9, which is formed by inactivating both HNH and RuvC endonuclease domains[32]. Because of the lack of nuclease activity in the dCas9, it has been utilized in many applications that exploited its DNA detection and localization and the ability to synthesize chimeric protein complexes when fused with different effector proteins [29][32][33]. These applications include genomic visualization via the fusion with fluorescent proteins, gene regulation through fusion with activators or repressors, alteration in epigenetic modifications through fusion with methyltransferases or deacetylases, and immunoprecipitation [32][33][34].
Base Editors
Base editors have recently emerged as efficient and specific genome engineering tools through the fusion of dCas9 with a catalytic domain that is capable of deaminating cytosine or adenine bases in the genome[35][36][37]. Two classes of DNA base editors have been described: cytosine base editors (CBEs) that convert a C•G base pair into a T•A base pair, and adenine base editors (ABEs) that convert an A•T base pair to a G•C base pair. Collectively, CBEs and ABEs can mediate all four possible transition mutations (C to T, A to G, T to C, and G to A)[36][38][39]. These base editors are considered effective genome engineering tools to potentially treat human genetic diseases, two-thirds of which are due to single-base alterations[40]. However, the applications using the base editor technique have several limitations, such as (i) the identification and isolation of cell populations that have been successfully edited, (ii) the generation of some off-target effects that occur when additional cytosines that are proximal to the target base get edited, and (iii) the location of the PAM sequence for the dCas9, which limits editing efficiency, as reported in plants [41][42].
FokI Endonuclease Fused to dCas9
To sum up the advantages of all possible gene-editing systems, researchers have postulated that using dCas9 as a binding module and fusing it to FokI will generate a new gene-editing platform with flexible RNA-guided specificity and controlled nuclease activity by FokI dimerization [43][44][45][46][47]. This new tool is referred to as FokI–dCas9 (fdCas9) or RNA-guided FokI nuclease (RFN) and was first introduced and validated in 2014 by Tsai and Guilinger and their colleagues[43][44]. fdCas9 has indeed reported almost complete elimination of any off-target effects resulting from using WT Cas9, monomeric, and dimeric sgRNA–nickase systems (Table 2)[44]. Additional work has also validated fdCas9 activity on human genes with known off-target sites by gene-editing detection techniques that include the T7 endonuclease I (T7EI) assay and sequence alignment [43][44][45][46][47]. Similarly, Fisicaro and his colleagues used unbiased whole genome sequencing and optimized bioinformatic analysis from pigs to further prove the high efficiency and specificity of fdCas9[48].
Table 2. Comparison between genomic editing activities of Nickases versus FokI–dCas9.
|
Nickases (pairs of D10 or H840) |
FokI-dCas9 |
|||
|
Structure |
||||
|
Existing forms |
Monomers |
Dimers |
Monomers |
Dimers |
|
Cleavage domain |
HNH or RuvC |
Pairs of HNH or pairs of RuvC |
FokI (But inactive) |
Pairs of FokI |
|
Obligate dimerization |
- |
No |
- |
Yes |
|
Spacer length |
- |
Up to 100bp |
- |
16-18bp or 26bp (depending on variant used) |
|
Target size |
17-20bp |
34-40bp + spacer length |
19-20bp |
38-40bp + spacer length |
|
Linker |
- |
- |
- |
Required to link the FokI domain |
|
Type of DNA damage |
Single strand nicks |
staggered double strand break |
No damage induced |
Double strands break |
|
Type of mutations |
Can induce point mutations |
Additions or deletions of >2bps |
Non-mutagenic |
Additions or deletions of >2bps |
|
Off-target effect (Compared to WT Cas9) |
Low-moderate |
Low |
Nearly non-existing |
Rare |
* Illustration figures created by BioRender.com
2. Engineering FokI–dCas9
To date, five constructs of fdCas9 have been engineered and published, aiming to optimize the activity, specificity, delivery, and feasibility of the system in order to encourage its usage. The varying parameters of compositions and characteristics of the designed constructs include the following:
2.1. FokI Fusion to dCas9
Previously published work from our group and others attempted FokI fusion to the C-terminal of dCas9 (dCas9-FokI) to mimic ZFN and TALEN architecture, but no editing activity was detected[43][44][45]. Active fdCas9 architecture was found to be through N-terminal FokI fusion (FokI–dCas9), positioning the FokI domain away from the PAM-interacting domain and closer to the cleavage site (Figure 1, Table 1)[43][44][45]. Presuming this effect is due to structural hindrance, a C-terminal fusion would work if the polypeptide linker between dCas9 and FokI is longer and more flexible in order to span the Cas9/sgRNA complex to reach the spacer site. However, even with such an alteration, the activity of the N-terminally fused FokI was superior[43][44][45].
2.2. Nuclear Localization Sequence (NLS)
The functional constructs required the presence of at least one NLS upstream of the dCas9[43][44][45][46][47]. Other constructs were engineered with multiple NLS copies at different terminus endings or within the domains to enhance nuclear delivery and the activity of fdCas9[44][45][46][47]. fdCas9 editing efficiency has been shown to be enhanced with the addition of 3× NLS at the C-terminal, in a similar way that was used to enhance editing activity using Cre and FLPe recombinases[45][49]. Increasing the copies of the N-terminal NLS has been shown to increase cellular penetration and nuclear delivery of WT SpCas9 due to the positive charge of the sequence. However, all fdCas9 constructs were engineered with only one N-terminal NLS (Figure 2) [43][44][45][46][47][50]. Thus, it could be worth increasing the N-terminal NLS copy number to enhance dCas9 nuclear delivery and activity.
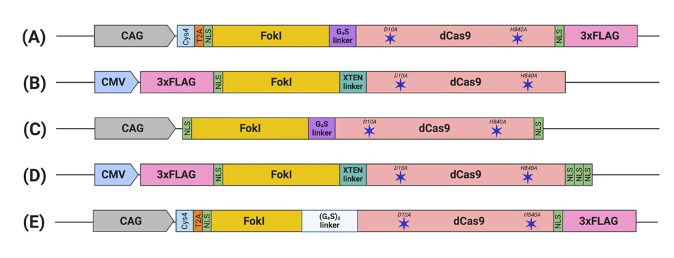
Figure 2. Schematic illustration of engineered FokI–dCas9 (fdCas9) constructs. Constructs are generated by (A) Tsai et al., (B) Guilinger et al., (C) Nakagawa et al., (D) Aouida et al., and (E) Havlicek et al. fdCas9 expression is driven by a strong constitutive human promoter (CMV and CAG). FokI endonuclease is fused to the N-terminal of the dead Cas9 (dCas9) [43–47]
2.3. Linker
Polypeptide linkers of different lengths and amino acid compositions were tested to amend the flexibility and accessibility of the system to the genomic sites for DNA cleavage[44]. Editing activity was resilient to changes in the linker between the N-terminal NLS and the FokI [44]. However, significant changes in activity were detected upon alterations to the linker between the FokI and dCas9[44]. Tsai et al. used the GlyGlyGlyGlyGlySer (GGGGS) amino acid linker, which showed a range of editing activity between 3% to 40% on various human genes[43]. In a subsequent work published by Guilinger et al., the authors tested 17 linkers with different lengths and amino acid compositions and reported varying editing activities with each linker, with the threshold activity observed with the 16-residue-long “XTEN” linker G [44], similar to the linker used in our generated construct[44][45]. Later, Havlicek et al. showed that the highest editing activity was detected using the long and flexible 25-residue-long (GGGGS)5 linker, even when compared to previously reported GGGGS and XTEN linkers[47]. Although the shorter GGGGS linker has induced similar editing activity with a spacer distance of 14 to 18 bp, an advantage of using the (GGGGS)5 linker is increasing activities at the spacer length to up to 26, 29, 37, 40, and/or 41 bp apart[47].
This entry is adapted from the peer-reviewed paper 10.3390/cells9112518
References
- Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science (80- ) 2014;346:1258096. doi:10.1126/science.1258096.
- [2] Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 1987;169:5429–5433. doi:10.1128/JB.169.12.5429-5433.1987.
- [3] Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci 2012;109:E2579–E2586. doi:10.1073/pnas.1208507109.
- [4] Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science (80- ) 2012;337:816–821. doi:10.1126/science.1225829.
- [5] Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 2013;31:233–239. doi:10.1038/nbt.2508.
- [6] Cong L, Ran FA, Cox D, Lin S, Barretto R, Hsu PD, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013;339:819–823. doi:10.1126/science.1231143.Multiplex.
- [7] Ding Y, Li H, Chen L-L, Xie K. Recent Advances in Genome Editing Using CRISPR/Cas9. Front Plant Sci 2016;7. doi:10.3389/fpls.2016.00703.
- [8] Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. Elife 2013;2. doi:10.7554/eLife.00471.
- [9] O’Geen H, Yu AS, Segal DJ. How specific is CRISPR/Cas9 really? Curr Opin Chem Biol 2015;29:72–78. doi:10.1016/j.cbpa.2015.10.001.
- [10] Karvelis T, Gasiunas G, Miksys A, Barrangou R, Horvath P, Siksnys V. crRNA and tracrRNA guide Cas9-mediated DNA interference in Streptococcus thermophilus. RNA Biol 2013;10:841–851. doi:10.4161/rna.24203.
- [11] Oakes BL, Nadler DC, Savage DF. Protein Engineering of Cas9 for Enhanced Function, 2014, p. 491–511. doi:10.1016/B978-0-12-801185-0.00024-6.
- [12] Li H, Yang Y, Hong W, Huang M, Wu M, Zhao X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct Target Ther 2020;5. doi:10.1038/s41392-019-0089-y.
- [13] Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 2013;31:397–405. doi:10.1016/j.tibtech.2013.04.004.
- [14] Ho B, Loh S, Chan W, Soh B. In Vivo Genome Editing as a Therapeutic Approach. Int J Mol Sci 2018;19:2721. doi:10.3390/ijms19092721.
- [15] Campa CC, Weisbach NR, Santinha AJ, Incarnato D, Platt RJ. Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat Methods 2019;16:887–893. doi:10.1038/s41592-019-0508-6.
- [16] Doyon Y, Vo TD, Mendel MC, Greenberg SG, Wang J, Xia DF, et al. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods 2011;8:74–79. doi:10.1038/nmeth.1539.
- [17] Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res 2015;75:3853–3864. doi:10.1158/0008-5472.CAN-14-3321.
- [18] Gautron A-S, Juillerat A, Guyot V, Filhol J-M, Dessez E, Duclert A, et al. Fine and Predictable Tuning of TALEN Gene Editing Targeting for Improved T Cell Adoptive Immunotherapy. Mol Ther - Nucleic Acids 2017;9:312–321. doi:10.1016/j.omtn.2017.10.005.
- [19] Lin Y, Cradick TJ, Brown MT, Deshmukh H, Ranjan P, Sarode N, et al. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res 2014;42:7473–7485. doi:10.1093/nar/gku402.
- [20] Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 2014;32:279–284. doi:10.1038/nbt.2808.
- [21] Kuscu C, Arslan S, Singh R, Thorpe J, Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol 2014;32:677–683. doi:10.1038/nbt.2916.
- [22] Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res 2014;24:132–141. doi:10.1101/gr.162339.113.
- [23] Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res 2013;41:9584–9592. doi:10.1093/nar/gkt714.
- [24] Ran FA, Hsu PD, Lin C-Y, Gootenberg JS, Konermann S, Trevino AE, et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013;154:1380–1389. doi:10.1016/j.cell.2013.08.021.
- [25] Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 2014;11:399–402. doi:10.1038/nmeth.2857.
- [26] Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8:2281–2308. doi:10.1038/nprot.2013.143.
- [27] Bothmer A, Phadke T, Barrera LA, Margulies CM, Lee CS, Buquicchio F, et al. Characterization of the interplay between DNA repair and CRISPR/Cas9-induced DNA lesions at an endogenous locus. Nat Commun 2017;8:13905. doi:10.1038/ncomms13905.
- [28] Wang B, Yan S. When and how to use nickases for efficient genome editing. Feb 14, 2018 2018. https://www.idtdna.com/pages/education/decoded/article/when-and-how-to-use-nickases-for-efficient-genome-editing.
- [29] Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-Guided Human Genome Engineering via Cas9. Science (80- ) 2013;339:823–826. doi:10.1126/science.1232033.
- [30] Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 2013;31:839–843. doi:10.1038/nbt.2673.
- [31] Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 2013;31:822–826. doi:10.1038/nbt.2623.
- [32] Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013;152:1173–1183. doi:10.1016/j.cell.2013.02.022.
- [33] Ma H, Tu LC, Naseri A, Huisman M, Zhang S, Grunwald D, et al. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat Biotechnol 2016;34:528–530. doi:10.1038/nbt.3526.
- [34] Anton T, Karg E, Bultmann S. Applications of the CRISPR/Cas system beyond gene editing. Biol Methods Protoc 2018;3:1–10. doi:10.1093/biomethods/bpy002.
- [35] Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017;551:464–471. doi:10.1038/nature24644.
- [36] Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016;533:420–424. doi:10.1038/nature17946.
- [37] Kurt IC, Zhou R, Iyer S, Garcia SP, Miller BR, Langner LM, et al. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat Biotechnol 2020. doi:10.1038/s41587-020-0609-x.
- [38] Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science (80- ) 2016;353:aaf8729–aaf8729. doi:10.1126/science.aaf8729.
- [39] Li C, Zong Y, Wang Y, Jin S, Zhang D, Song Q, et al. Expanded base editing in rice and wheat using a Cas9-adenosine deaminase fusion. Genome Biol 2018;19:1–9. doi:10.1186/s13059-018-1443-z.
- [40] Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science (80- ) 2014;343:84–87. doi:10.1126/science.1247005.
- [41] Jin S, Zong Y, Gao Q, Zhu Z, Wang Y, Qin P, et al. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science (80- ) 2019:eaaw7166. doi:10.1126/science.aaw7166.
- [42] Mishra R, Joshi RK, Zhao K. Base editing in crops: current advances, limitations and future implications. Plant Biotechnol J 2020;18:20–31. doi:10.1111/pbi.13225.
- [43] Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 2014;32:569–576. doi:10.1038/nbt.2908.
- [44] Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol 2014;32:577–582. doi:10.1038/nbt.2909.Fusion.
- [45] Aouida M, Eid A, Ali Z, Cradick T, Lee C, Deshmukh H, et al. Efficient fdCas9 synthetic endonuclease with improved specificity for precise genome engineering. PLoS One 2015;10:1–16. doi:10.1371/journal.pone.0133373.
- [46] Nakagawa Y, Sakuma T, Sakamoto T, Ohmuraya M, Nakagata N, Yamamoto T. Production of knockout mice by DNA microinjection of various CRISPR/Cas9 vectors into freeze-thawed fertilized oocytes. BMC Biotechnol 2015;15:1–10. doi:10.1186/s12896-015-0144-x.
- [47] Havlicek S, Shen Y, Alpagu Y, Bruntraeger MB, Zufir NBM, Phuah ZY, et al. Re-engineered RNA-Guided FokI-Nucleases for Improved Genome Editing in Human Cells. Mol Ther 2017;25:342–355. doi:10.1016/j.ymthe.2016.11.007.
- [48] Fisicaro N, Salvaris EJ, Philip GK, Wakefield MJ, Nottle MB, Hawthorne WJ, et al. FokI-dCas9 mediates high-fidelity genome editing in pigs. Xenotransplantation 2020;27:1–6. doi:10.1111/xen.12551.
- [49] Andreas S, Schwenk F, Küter-Luks B, Faust N, Kühn R. Enhanced efficiency through nuclear localization signal fusion on phage ΦC31-integrase: Activity comparison with Cre and FLPe recombinase in mammalian cells. Nucleic Acids Res 2002;30:2299–2306. doi:10.1093/nar/30.11.2299.
- [50] Staahl BT, Benekareddy M, Coulon-Bainier C, Banfal AA, Floor SN, Sabo JK, et al. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat Biotechnol 2017;35:431–434. doi:10.1038/nbt.3806.