Ataxia-Telangiectasia (A-T) is a rare autosomal recessive disorder, characterized by a progressive cerebellar neurodegeneration, immunodeficiency, infertility, and cancer predisposition, with high incidence of leukemia and lymphoma.
- bone marrow transplantation
- ATM
- Ataxia telangiectasia
1. Introduction
A-T is caused by a deficiency of ATM (Ataxia-Telangiectasia Mutated) protein, encoded by a gene at chromosome 11q23 in humans[1]. The ATM protein belongs to the family of phosphatidylinositol 3-kinases that share homologies in the catalytic domain. The kinase functions as a guardian of genome integrity acting on cell-cycle checkpoints, apoptosis, gene expression, and DNA repair after DNA double-strand breaks (DSBs)[2]. DSBs are generated in the cells by exogenous and endogenous insults such as ionizing radiation, chemical reagents, replication, DNA demethylation, meiotic recombination, and during the generation of antibody diversity in the immune system.
ATM is recruited and activated by the MRN protein complex at DSBs. A phosphorylation- acetylation cascade sustains ATM activation by c-Abl tyrosine kinase and chromatin-bound TIP60 acetyltransferase[3]. Following activation, ATM acts as a DSB transducer activating a plethora of downstream effectors and orchestrating the DNA damage response[2].
2. Ataxia-Telangiectasia Syndrome: Diagnosis and Current Treatments
Ataxia-telangiectasia (A-T) is a complex disorder that affects 1: 100 000 live births and it is clinically represented by a wide variability in patients[4][5]. Affected individuals can show early childhood-onset progressive cerebellar decline (ataxia), telangiectasia, predisposition to progressive bronchopulmonary disease, and cancer development, especially of lymphoid origin. A-T patients are also characterized by immunodeficiency with hypoplasia or agenesis of the thymus gland, clinical and cellular hypersensitivity to ionizing radiation and radiomimetic drugs. Usually, they show premature aging with progeria-type hair and skin changes, growth retardation, endocrine abnormalities, gonadal atrophy, choreoathetosis with dystonic posturing of hands and feet, osteoporosis, insulin-resistant diabetes. Mental retardation is not commonly seen in A-T patients[4][6][7].
The A-T diagnosis is made by association of clinical features and specific laboratory findings, such as cerebellar atrophy on magnetic resonance imaging and lack of voluntary movement coordination, including gait abnormality, elevated serum alpha-fetoprotein (AFP) levels after two years of age, low serum levels of immunoglobulins (IgA, IgG, IgG subclasses and IgE), and lymphopenia[4][5][6]. The diagnosis confirmation is done by the assessment of absence or deficiency in the ATM protein and/or ATM kinase activity in cultured cell lines from lymphocytes or skin biopsies and the detection of mutations in the ATM gene[4].
Since there are no cures for A-T yet, the management and treatment for this syndrome are symptomatic and supportive, aiming to give patients a more comfortable life and to alleviate the symptoms[4][8]. As A-T is a multisystem disease, a “multidisciplinary therapy” is required to interfere with the progressive neurodegeneration, correct the immunodeficiency, diminish the proneness to cancer, and alleviate bronchial complications[8][9]. Immunoglobulin replacement therapy is recommended only to patients with life-threatening or frequently recurring infections[7]. For the other patients, it is recommended the use of inactivated vaccines as normal childhood vaccination routine[4][9]. Since the generation of excessive free radicals creates life-threatening DNA lesions, antioxidant therapies are recommended for A-T patients, although there are no published clinical trials showing therapeutic advantages[5][10]. It has also been suggested that a wide-spectrum of neuroprotective factors such as insulin-like growth factor 1 (IGF-1) could be beneficial in cerebellar ataxia treatments[11], and a clinical trial is being performed at the “Johann Wolfgang Goethe University Hospital”, Frankfurt, Germany (NCT01052623). A clinical trial showed that the administration of growth hormone in A-T patients increased the height of patients, but did not affect lymphocytes and ataxia subsets[12]. Steroid hormones show powerful anti- inflammatory action, capable of crossing the blood−brain barrier, and for this reason they are used to treat the neurological symptoms of A-T as reported from case reports[13][14]. Indeed, to date, the specific role of steroids has not been established and the clinical trials have not yet conclusively shown benefit.
A clinical guidance for diagnosis and treatment of Ataxia Telangiectasia in children has been created (http://www.Atsociety.org.uk/data/files/William/A-T_Clinical_Guidance_Document_Final.pdf) and since 2016 the Global A-T Family Data Platform (https://www.atfamilies.org/) and the A-T International registry (http://www.atregistry.eu/) are available for patients, their parents, scientists, and physicians.
3. Bone Marrow/Hematopoietic Stem Cell Transplantation as a Therapeutic Approach
Primary immunodeficiency of Ataxia-Telangiectasia might ameliorate after Bone Marrow (BM)/Hematopoietic Stem Cell (HSC) Transplantation (Figure 1), however so far very few case reports were published. In the early 1970s a boy with A-T and IgA deficiency was found with transient improvements in the immune system and normal IgA level several months after an infusion of bone marrow cells from a compatible healthy sibling[15]. However, no evidence of chimerism was detected in his peripheral blood lymphocytes and bone marrow cells. More recently, a Turkish child of 22 months of age, whose A-T diagnosis was made postmortem, was transplanted for a misdiagnosed immunodeficiency and died 8 months after, due to hepatic failure and encephalopathy[16]. In 2013 it was reported the first long-term survival of an A-T patient after HSCT at the age of 3 years old, from his HLA-identical sibling[17]. A modified protocol of the conditioning used in the Fanconi anemia disease (GEFA02;[18]) was employed to prevent graft rejection. A follow-up published 8 years after transplantation[19], reported that the patient remained in leukemia remission, he was attending primary school but showed coordination deficits and when he was around age 12 he lost the ability to walk, becoming wheelchair-bound. More recently, other 2 children (12 months old and 23 months old) presenting the classic version of A-T were conditioned using a modified Fanconi anemia protocol (GEFA03)[19]. They survived the procedure without significant adverse effects, showing mild mucosal toxicities and maintaining oral feeding ability throughout the transplant period. The patients were engrafted and lymphocyte reconstitution was observed. Both children are in their first decade of life and exhibit mild neurologic deficits, but the 12-months old treated children head magnetic resonance revealed signs of cerebellar atrophy. No improvement was observed in growth and weight gain after HSCT, which remained poor, however, gastrostomy tube feeding was not considered by patients’ families. The next reported case of BMT in an A-T patient was published in 2016. A 13-year-old boy who also developed EBV-positive non-Hodgkin lymphoma was cured by chemotherapy associated with allogenic-matched sibling HSCT (allo-HSCT)[20]. The conditioning used was based on the same Fanconi anemia protocol (GEFA02). This patient tolerated the therapy and achieved remission, but he did not tolerate the conditioning well, developing life-threatening toxicity in several organ systems. Bakhtiar et al., 2018[21] reported a 6-year follow-up of a pre-emptive allogenic HSCT in a 4-year-old boy with A-T. At the age of 3 he presented with upper respiratory infections and analysis revealed very low naïve T cells, absence of IgA and low IgG2 and IgG4. The patient also developed granulomas in the hands and elbow. The bone marrow from an HLA identical sibling was used to perform HSCT and a reduced intensity conditioning (RIC) regimen. Regarding the treatment in this patient, no signs of acute toxicity were observed and it provided complete immunological reconstitution along with the remission of granulomas without any need for immunosuppression or immunoglobulin replacement. The patient gained height and weight, exhibited a milder progression of ataxic symptoms compared to age-matched A-T patients.
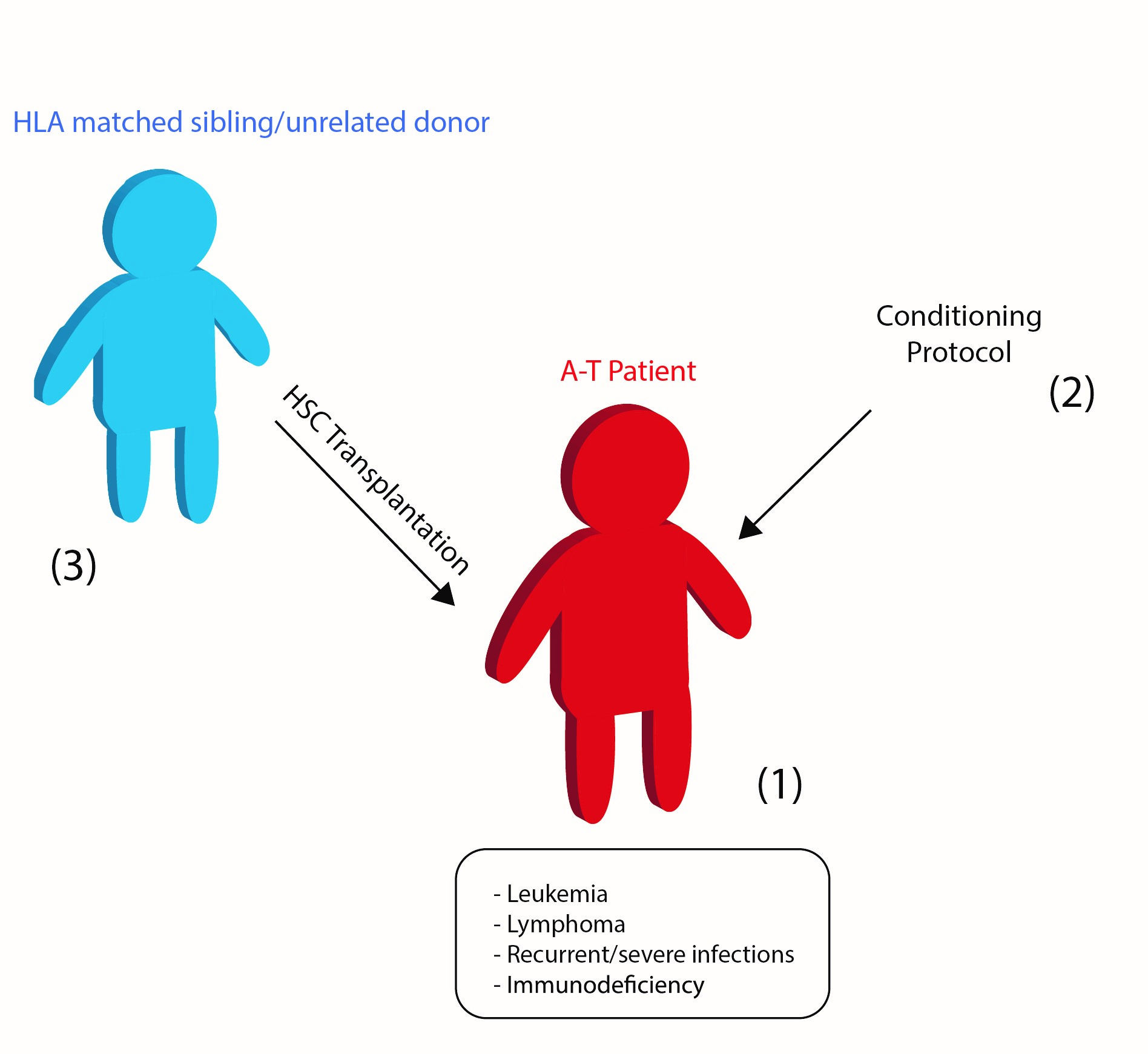
Figure 1 - The A-T patient with particular clinical profile (1) and a specific conditioning protocol (2) can be subjected to HSC transplantation with cells collected from a HLA- matched donor (3).
4. Conclusions
To date, only a few A-T children have been transplanted with HSCs because of the high risk of mortality due to excessive toxicities and the ethical eligibility to perform HSCT on A-T patients remains controversial. However, a very small and highly selected proportion of A-T patients, with clinical complications such as recurrent infections, severe manifestations of immune deficiency and children after cancer therapy, have been subjected to BMT in particular allo-HSCT, that induced T lymphocytes and immunoglobulins restoration and prevented severe infections or signs of lung disease, at least in the short period of the follow-up. BMT was not able to evidently ameliorate the neurological symptoms and disability, which for most patients are the most important and quality-of-life limiting aspects of the disease, nor to rescue body growth and the serum AFP levels of A-T patients. A longer period of observation is necessary to understand the impact of HSCT on lymphoma and leukemia predisposition. Moreover, since allo-HSCT in A-T is an invasive and risky treatment, transplant guidelines are absolutely required before considering pilot studies and randomized controlled trials.
This entry is adapted from the peer-reviewed paper 10.3390/cancers12113207
References
- 1. Lange, E.; Borresen, A.L.; Chen, X.; Chessa, L.; Chiplunkar, S.; Concannon, P.; Dandekar, S.; Gerken, S.; Lange, K.; Liang, T.; et al. Localization of an ataxia-telangiectasia gene to an ~500-kb interval on chromosome 11q23.1: Linkage analysis of 176 families by an international consortium. Am. J. Hum. Genet. 1995, 57, 112–119.
- Yosef Shiloh; ATM and related protein kinases: safeguarding genome integrity. Nature Reviews Cancer 2003, 3, 155-168, 10.1038/nrc1011.
- Andrew N. Blackford; Stephen D. Jackson; ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Molecular Cell 2017, 66, 801-817, 10.1016/j.molcel.2017.05.015.
- Cynthia Rothblum-Oviatt; Jennifer Wright; Maureen A. Lefton-Greif; Sharon A. McGrath-Morrow; Thomas O. Crawford; Howard M. Lederman; Ataxia telangiectasia: a review. Orphanet Journal of Rare Diseases 2016, 11, 1-21, 10.1186/s13023-016-0543-7.
- Gatti, R.; Perlman, S. Ataxia-Telangiectasia. In; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; Seattle (WA), 2016
- Chun, H.H.; Gatti, R.A. Ataxia-telangiectasia, an evolving phenotype. DNA Repair (Amst). 2004, 3, 1187–1196
- Perlman, S.L.; Boder (deceased), E.; Sedgewick, R.P.; Gatti, R.A. Ataxia-telangiectasia; 1st ed.; Elsevier B.V., 2012; Vol. 103
- Martin F. Lavin; Nuri Gueven; Stephen Bottle; Richard A. Gatti; Current and potential therapeutic strategies for the treatment of ataxia-telangiectasia. British Medical Bulletin 2007, 81–82, 129-147, 10.1093/bmb/ldm012.
- Nienke J.H. Van Os; Anne F. M. Jansen; Marcel Van Deuren; Asgeir Haraldsson; Nieke T.M. Van Driel; Amos Etzioni; Michiel Van Der Flier; Charlotte A. Haaxma; Tomohiro Morio; Amit Rawat; et al. Ataxia-telangiectasia: Immunodeficiency and survival. Clinical Immunology 2017, 178, 45-55, 10.1016/j.clim.2017.01.009.
- Mark Ambrose; R. A. Gatti; Pathogenesis of ataxia-telangiectasia: the next generation of ATM functions. Blood 2013, 121, 4036-4045, 10.1182/blood-2012-09-456897.
- A. M. Fernandez; E. M. Carro; C. Lopez-Lopez; Ignacio Torres Alemán; Insulin-like growth factor I treatment for cerebellar ataxia: Addressing a common pathway in the pathological cascade?. Brain Research Reviews 2005, 50, 134-141, 10.1016/j.brainresrev.2005.05.003.
- Sandra Woelke; Eva Valesky; Shahrzad Bakhtiar; Helena Pommerening; L. M. Pfeffermann; Ralf Schubert; S. Zielen; Treatment of Granulomas in Patients With Ataxia Telangiectasia. Frontiers in Immunology 2018, 9, 1–8, 10.3389/fimmu.2018.02000.
- Giardino, G.; Fusco, A.; Romano, R.; Gallo, V.; Maio, F.; Esposito, T.; Palamaro, L.; Parenti, G.; Salerno, M.C.; Vajro, P.; et al. Betamethasone therapy in ataxia telangiectasia: Unraveling the rationale of this serendipitous observation on the basis of the pathogenesis. Eur. J. Neurol. 2013.
- Buoni, S.; Zannolli, R.; Sorrentino, L.; Fois, A. Betamethasone and improvement of neurological symptoms in ataxia-telangiectasia. Arch. Neurol. 2006.
- R H Buckley; Bone marrow and thymus transplantation in ataxia-telangiectasia.. Birth defects original article series 1975, 11, 421–424, .
- Sujal Ghosh; Friedhelm R. Schuster; Vera Binder; Tim Niehues; Stephan E. Baldus; Peter Seiffert; Hans-Jürgen Laws; Arndt Borkhardt; Roland Meisel; Fatal Outcome Despite Full Lympho-Hematopoietic Reconstitution After Allogeneic Stem Cell Transplantation in Atypical Ataxia Telangiectasia. Journal of Clinical Immunology 2012, 32, 438-440, 10.1007/s10875-012-9654-7.
- Marek Ussowicz; J Musiał; E Duszeńko; O Haus; K Kałwak; Long-term survival after allogeneic-matched sibling PBSC transplantation with conditioning consisting of low-dose busilvex and fludarabine in a 3-year-old boy with ataxia-telangiectasia syndrome and ALL. Bone Marrow Transplantation 2012, 48, 740-741, 10.1038/bmt.2012.207.
- M. M. Chao; W. Ebell; P. Bader; R. Beier; B. Burkhardt; T. Feuchtinger; R. Handgretinger; H. Hanenberg; U. Koehl; C. Kratz; et al. Consensus of German Transplant Centers on Hematopoietic Stem Cell Transplantation in Fanconi Anemia. Klinische Pädiatrie 2015, 227, 157-165, 10.1055/s-0035-1548841.
- Marek Ussowicz; Elżbieta Wawrzyniak-Dzierżek; Monika Mielcarek-Siedziuk; Małgorzata Salamonowicz; Jowita Frączkiewicz; Blanka Rybka; Renata Ryczan-Krawczyk; Krzysztof Kałwak; Allogeneic Stem Cell Transplantation after Fanconi Anemia Conditioning in Children with Ataxia-Telangiectasia Results in Stable T Cell Engraftment and Lack of Infections despite Mixed Chimerism. Biology of Blood and Marrow Transplantation 2018, 24, 2245-2249, 10.1016/j.bbmt.2018.07.001.
- R. Beier; K-W Sykora; W. Woessmann; B. Maecker-Kolhoff; M. Sauer; H. H. Kreipe; T. Dörk-Bousset; C. Kratz; M. Lauten; Allogeneic-matched sibling stem cell transplantation in a 13-year-old boy with ataxia telangiectasia and EBV-positive non-Hodgkin lymphoma. Bone Marrow Transplantation 2016, 51, 1271-1274, 10.1038/bmt.2016.93.
- Shahrzad Bakhtiar; Sandra Woelke; Sabine Huenecke; Matthias Kieslich; Alexander Malcolm Taylor; Ralf Schubert; Stefan Zielen; Peter Bader; Pre-emptive Allogeneic Hematopoietic Stem Cell Transplantation in Ataxia Telangiectasia. Frontiers in Immunology 2018, 9, 2495, 10.3389/fimmu.2018.02495.