Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Others
Integrated CO2 capture and utilization aims to capture CO2 from gas streams and other emission sources and convert it into valuable chemicals or energy sources. The key is to find the match between the CO2 separation process and the CO2 utilization process, including the temperature and pressure, etc. CO2 can be captured via physical or chemical absorption depending on the interaction between CO2 and the absorbent.
- carbon neutrality
- CO2 capture
- CO2 conversion
- integration
1. Introduction
Carbon dioxide capture, utilization, and storage (CCUS) are increasingly gaining global attention. The challenge is to meet the energy demand while balancing CO2 emissions. Several solutions have been proposed to reduce the CO2 emission, namely, increasing the utilization of eco-friendly energy sources, such as wind and solar energy, to improve the energy efficiency. However, the advancement of such technologies is currently still limited and can be an optimal option in the long-term. At present times, the CCUS technology is more effective and can be a short-term alternative.
CO2 can be captured using various technologies, such as amine absorption, porous materials adsorption and membrane separation. Since CO2 itself is a carbon source, it can be converted into valuable chemicals via dry methane reforming (DMR), CO2 hydrogenation, reverse water–gas shift reaction, etc. There are excellent reviews on both the technologies [1,2,3]. However, the traditional CO2 capture and utilization processes are separated, which inevitably increases the transportation cost. To reduce or even eliminate such cost, the integration of CO2 capture and utilization is greatly desirable.
2. Integration of CO2 Capture and Utilization
Integrated CO2 capture and utilization aims to capture CO2 from gas streams and other emission sources and convert it into valuable chemicals or energy sources. The key is to find the match between the CO2 separation process and the CO2 utilization process, including the temperature and pressure, etc.
2.1. Integration of CO2 Absorption and Conversion
CO2 can be captured via physical or chemical absorption depending on the interaction between CO2 and the absorbent. The former mainly utilizes the solubility of each gas component in the solvent whereas the latter involves chemical reactions between CO2 and the absorbent. Chemical absorption is mostly employed with organic amine, hot potash, and liquid ammonia solvents considering its easy operation and mild working conditions [4]. Accordingly, it can be integrated with CO2 hydrogenation for the production of formic acid or methanol.
Amine-based CO2 capture and conversion integrated reactors consist of a series of amine sorbents and metal ions that form a pincer complex, such as pincerpentaethylenehexamine (PEHA), pyrrolizidine, N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDTA) and polyethyleneimine (PEI). These are coupled with metal pincer-based homogeneous catalysts [5,6,7,8]. Due to the presence of multiple amine sites [9], PEI can be used as a superior CO2 absorber [10], which can absorb both high and low concentrations of CO2 [11]. Li et al. [12] used PEI/RhCl3·3H2O/CyPPh2 to capture CO2 and convert it in situ (Figure 1a). PEI absorbs CO2 from air in an ethylene glycol solution containing PEI and converts it into amino formate esters and alkyl carbonate esters. CO2 is hydrogenated in situ to form formate salts (TON = 260) in the presence of RhCl3-3H2O/CyPPh2, demonstrating the first in situ CO2 capture and conversion to formate. Multifunctional materials can be synthesized by modifying the PEI backbone with iminophosphine ligand functionality and subsequently metallizing it with Ir precursors. About 65% of the available primary amines on PEI can be modified to form iminophosphine/Ir (PN/Ir) for balanced CO2 capture and conversion, resulting in higher formic acid yields. PEI with relatively lower molecular weight has better CO2 capture ability and catalytic activity (Figure 1b) [13]. Kothandaraman et al. [14] reported, for the first time, a green and simplified method for in situ conversion of captured CO2 to formate in aqueous media in the presence of Ru- and Fe-based pincer complexes without excess alkali, with yields of up to 95% of the formate.
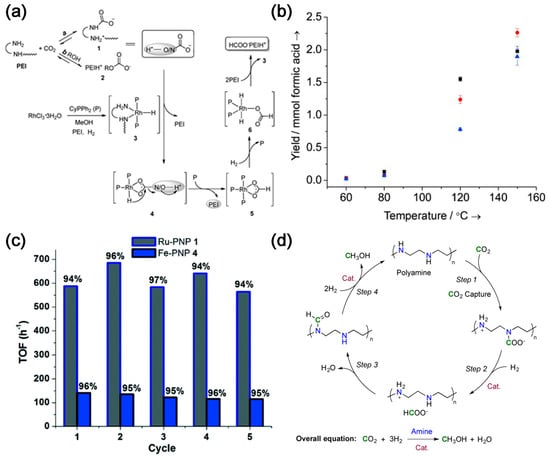
Figure 1. (a) Proposed pathways of carbon capture and subsequent hydrogenation of the captured CO2 [12]; (b) Formic acid yields in the hydrogenation of CO2 catalyzed by the PEI–PN/Ir materials as a function of temperature and MW of PEI for PEI600–PN/Ir (▪), PEI1800–PN/Ir (•), and PEI25 000–PN/Ir (▴) [13]. (c) Multiple recycling of the catalyst in biphasic reaction mixture. Yield (%) of formate is relative to the amount of CO2 captured. Ru–PNP 1: Cat 1 = 10 μmol, T = 55 °C, H2 = 50 bar, 17.2 mmol diazabicyclo[2.2.2]octane (DABCO) + 3 mL H2O (CO2 captured each cycle = 15 mmol), 3 mL additional H2O–4 mL 2-Methyltetrahydrofuran (2–MeTHF) added for hydrogenation study. Fe–PNP 4: Cat 4 = 20 μmol, T = 55 °C, H2 = 50 bar, 17.2 mmol DABCO + 3 mL H2O (CO2 captured each cycle = 15 mmol), 3 mL additional H2O–4 mL 2–MeTHF added for hydrogenation study [14]. (d) Proposed reaction sequence for CO2 capture and in situ hydrogenation to CH3OH using a polyamine [5].
In addition to formate, the captured CO2 can be converted into methanol. In 2015, Rezayee et al. [15] prepared methanol by tandem CO2 capture and in situ conversion of dimethylamine with homogeneous ruthenium complexes under basic conditions. Dimethylamine can react with CO2 and inhibit the formation of formic acid. The conversion of CO2 is >95% (Figure 1c). Moreover, Kothandaraman et al. [5] first proposed and demonstrated a system for capturing CO2 from air (~400 ppm) and converting it in situ (Figure 1d), which consisted of PEHA and Ru–PNP complex, with a methanol yield of 79%. Integrated systems for CO2 capture and conversion into methanol are still uncommon. The major obstacle is the harsh reaction conditions to produce methanol, which involves a high-pressure gas-phase catalyst reactor at relatively low temperatures of 200–300 °C and high pressures of 50–100 atm.
The amine-based materials offer the potential to capture and convert CO2 under mild conditions. Due to the relatively high CO2 capture capacity of PEI, it can directly capture CO2 from the air, which also removes the limitation of constructing CO2 capture equipment. However, the high selectivity of some materials to carbon dioxide poses a challenge for the regeneration of amines. Furthermore, the toxicity and corrosiveness of amine solvents limit their industrial application. To drive future research, it is essential to explore systems that are highly efficient and recyclable, enabling CCU to establish a reliable foundation for industrial applications.
2.2. CO2 Adsorption and Conversion Integration
Similar to absorption, adsorption separation can also be divided into chemisorption and physisorption according to the interaction between CO2 and the adsorbate, with the former forming covalent bonds whilst the latter forms bond with electrostatic attraction and van der Waals forces.
Porous organic polymers (POPs) are a series of new two- or three-dimensional networked polymeric materials formed by covalent bonding of organic small molecule substrates through specific chemical reactions, usually with microporous, mesoporous or multistage pore structures. They have promising applications in separation, sensor and catalysis [16]. The ionic porous organic polymers (IPOPs) are generally classified as porous organic materials, whose backbone typically includes anions or cations. They can be divided into IPOPs with cationic moieties, IPOPs with anionic moieties, and IPOPs with zwitterionic moieties. Common cationic moieties include imidazolium, pyridinium, viologen, and quaternary phosphonium, whereas more common anionic moieties include tetrakis(phenyl)borate and tris(catecholate) phosphate. The inclusion of these ionic moieties in porous materials can enhance their CO2 capture capacity and catalyze the in situ CO2 conversion.
In 2011, tetrakis(4-ethynylphenyl)methane and diiodoimidazolium salts were used to prepare tubular microporous organic via Sonogashira coupling reaction networks bearing imidazolium salts (T-IM) (Figure 2a). The material, with a microporous structure of specific surface area (620 m2 g−1), shows good catalytic activity towards the conversion of CO2 to cyclic carbonates [17]. Wang et al. [18] utilized the Friedel–Crafts reaction to synthesize imidazole-based IPOPs. The specific surface area can reach up to 926 m2 g−1; however, its CO2 capture capacity is only 14.2 wt%. Nevertheless, the polymer exhibits good stability and repeatability. Sun et al. [19] demonstrated, for the first time, the capture and in situ conversion of CO2 into cyclic carbonates under relatively mild room temperature conditions using a metal-free solvent followed by a heterogeneous catalytic system. The effect of halogen anions (Cl, Br, and I) and quaternary phosphonium cations on the catalytic activity was investigated. The catalytic activity follows the order Cl− > Br− > I− (Figure 2b), because the rate-controlling step of the reaction is ring opening by anion attack on the epoxide [20].
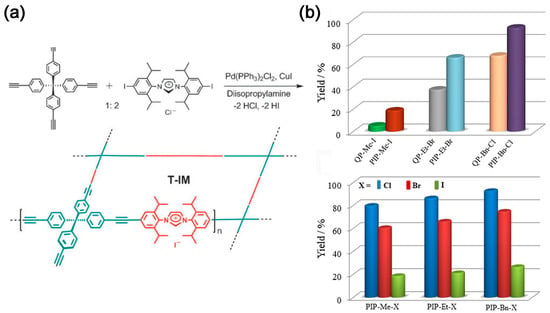
Figure 2. (a) Preparation of porous organic networks bearing imidazolium salts [17]; (b) Yields of chloropropene carbonate from the cycloaddition of epichlorohydrin and CO2 catalyzed by PIPs with corresponding QPs and PIP-Me-X, PIP-Et-X, and PIP-Bn-X (X = Cl, Br, and I). Reaction conditions: epichlorohydrin (1.0 g, 10.9 mmol), catalyst (0.05 mmol, based upon the quaternary phosphonium salt), 323 K, CO2 (ambient pressure), and 24 h [19].
In addition to IPOP, the porphyrin-based organic polymer (POP-TPP) can also be used. Xiao et al. [21] synthesized a hierarchically porous organic polymer (POP-TPP) by polymerizing vinyl-functionalized tetraphenylporphyrin monomer, and then metalated it with different metals (Co3+, Zn2+, and Mg2+). The resulting heterogeneous catalysts have rich active sites and exhibit higher activity than the homogenous Co/TPP catalyst at relatively low CO2 concentrations, primarily due to the favorable enrichment of CO2 in the porous structure (micropores and nanochannels) of Co/POP-TPP. The TOF of the catalysts decreases in the following order: Co/POP-TPP (436 h−1) > Zn/POP-TPP (326 h−1) > Mg/POP-TPP (171 h−1) (Table 1). Later, a series of high surface area hollow tubular metal (Al, Co, Fe, and Mn) porphyrin-based hypercrosslinked polymers (HCP) were synthesized via Friedel–Crafts alkylation reactions. Al-HCP can catalyze the formation of propylene carbonate with a selectivity of approximately 99% after only 1 h with 2.0 mol% TBAB catalyst [22].
Table 1. Cycloaddition of epichlorohydrin with CO2 to form cyclic carbonate over various catalysts at 29 °C [21].
| Entry | Catalysts | Additives | Time (h) | Conv. (%) a | Select. (%) a |
|---|---|---|---|---|---|
| 1 b | POP-TPP | n-Bu4NBr | 24 | 52.1 | >99.0 |
| 2 c | Co/POP-TPP | n-Bu4NBr | 24 | 95.6 | 99 |
| 3 c | Co/TPP | n-Bu4NBr | 24 | 97.5 | 99 |
| 4 | Co/POP-TPP | None | 24 | 9.7 | 99 |
| 5 | Co/TPP | None | 24 | 18.5 | 99 |
| 6 | None | n-Bu4NBr | 24 | 34 | 99 |
| 7 d | Co/POP-TPP | n-Bu4NBr | 96 | 96.1 | 99 |
| 8 e | Co/POP-TPP | n-Bu4NBr | 24 | 88.9 | 99 |
| 9 f | Zn/POP-TPP | n-Bu4NBr | 24 | 93.2 | >99.0 |
| 10 f | Zn/TPP | n-Bu4NBr | 24 | 93.5 | >99.0 |
| 11 g | Mg/POP-TPP | n-Bu4NBr | 24 | 80.5 | >99.0 |
| 12 g | Mg/TPP | n-Bu4NBr | 24 | 99.3 | >99.0 |
| 13 h | Co/POP-TPP | n-Bu4NBr | 24 | 93.6 | 99 |
Note: a Determined using GC analysis. b 48 mg of POP-TPP was used. c 50 mg of Co/POP-TPP (1.6 mg Co) or 19 mg of Co/TPP (1.6 mg Co) was used. d Co/POP-TPP catalyst (12.5 mg, 0.4 mg Co). e 50 mg of n-Bu4NBr. f 50 mg of Zn/POP-TPP (2.3 mg Zn) or 23.5 mg of Zn/TPP (2.3 mg Zn) was used. g 50 mg of Mg/POP-TPP (1.4 mg Mg) or 37.0 mg of Mg/TPP (1.4 mg Mg) was used. h Recycled for 18 times.
Metal–organic frameworks (MOFs) are porous crystalline materials formed by coordination between metal ions or metal clusters and organic ligands, which are characterized by high porosity, high surface area, tunable pore size and geometric configuration, and functionalizable pore surface [23]. Consequently, they can be employed for CO2 capture and utilization. The imidazolium-based poly(ionic liquid)s (denoted as polyILs) and ortho-divinylbenzene were used as cross-linking agents to polymerize in the pores of MIL-101 (Figure 3a), resulting in polyILs@MIL-101 materials with dual functions of CO2 capture and conversion [24]. For MOFs materials, ordered nanochannels can effectively promote the enrichment of CO2 at the active sites and accelerate the CO2 conversion; Lewis basic sites (LBSs) can supply electrons to activate CO2 [25,26]. In order to restore the real CO2 capture process, a CO2 capture and in situ conversion system was designed by simulating the flue gas feed, where the lanthanide (III) complex of 1-vinylimidazole (Vim) was immobilized with DUT-5 by a combination of ligand and in situ polymerization (Figure 3b). This work opens up a general pathway for future research and validates the effectiveness of MOFs for practical applications in CO2 capture and conversion [27].
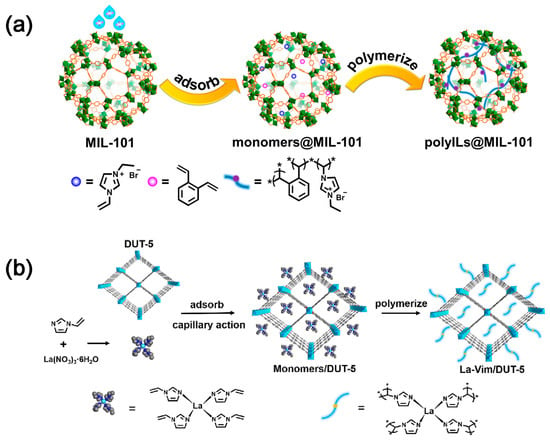
Although the integrated materials have demonstrated good CO2 capture and enrichment capabilities, their performance under the real industrial exhaust emissions that contain acidic gases, such as SOx, NOx, and steam, remain to be explored.
2.3. CO2 Electrochemical Membrane Separation and Conversion Integration
Electrochemical membrane separation is a technology that achieves gas separation through electrochemical reaction. The membrane can be mixed oxide ion-carbonate conductor (MOCC), such as Y0.16Zr0.84O2-δ-molten carbonate (YSZ-MC), Ce0.8Sm0.2O1.9-MC (SDC-MC), Ce0.9Gd0.2O1.9-MC (GDC-MC), and Bi1.5Y0.3Sm0.2O3-δ-MC (BYS-MC); mixed electron-carbonate conductor (MECC), such as stainless steel-MC (SS-MC), Ag-MC, and Ni-MC; or mixed oxide ion-electron-carbonate conductor (MOECC), such as La0.6Sr0.4Co0.8Fe0.2O3-δ-MC (LSCF-MC), La0.5Sr0.5Fe0.8Cu0.2O3-δ-MC (LSFCu-MC) and NiO-SDC-MC based on the charge carriers [2]. Generally, the carbonates (Li-Na-K-based) show high electrical conductivity and low viscosity only at relatively high temperature, e.g., >600 °C. Accordingly, it can be well integrated with the high temperature CO2 utilization processes, such as DMR and reverse water–gas shift (RWGS) (Table 2).
Lin and co-workers first modeled the high temperature tube shell membrane reactor for separation and utilization of CO2 from the flue gas and for simultaneous production of syngas using DMR. The membrane reactor is highly efficient for CCUS. The CH4 conversion of 48.1% and an average CO2 permeation flux of 1.52 mL cm−2 min−1 can be obtained at 800 °C with a 75 µm thick membrane and CH4 space velocity of 3265 h−1 [28]. Later, Anderson et al. [29] experimentally studied the integration of CO2 separation and DMR with LSCF-MC membrane and Ni/γ-alumina catalyst. The CO2 permeation rate above 750 °C matches the reaction rate of DMR catalyst, and the order of syngas production activity is blank system < LSCF combustion catalyst < Ni/γ-alumina reforming catalyst. The conversion of CO2 and CH4 is 88.5 and 8.1%, respectively, generating a syngas H2:CO ratio of about 1 [30]. To improve the performance, Zhang et al. [31] employed MOCC membrane (GDC-MC) (Figure 4a), and NMP (Ni-MgO-1 wt% Pt) and LNF (LaNi0.6Fe0.4O3-δ) catalysts. The yield of H2 and CO and the conversion of CH4 in the reactor with NMP catalyst is higher than that with LNF catalyst (Figure 4b,c), while the reactor with LNF catalyst shows better anti-coking performance and no significant degradation within 200 h. In addition, Zhang et al. [32] investigated the MECC membrane reactor (Ag-MC) coupled with dry-oxy methane reforming (DOMR) over an NMP catalyst. The CH4 conversion is >82% and is stable over 115 h at 800 °C (Figure 4d). Similar reactor models can be applied to integrate CO2 separation and oxidative dehydrogenation of ethane (ODHE) to prepare ethylene [33]. CO2 can react with H2 to make the reaction of ethane dehydrogenation forward, which can increase the selectivity of O2-ODHE to produce ethylene [33].
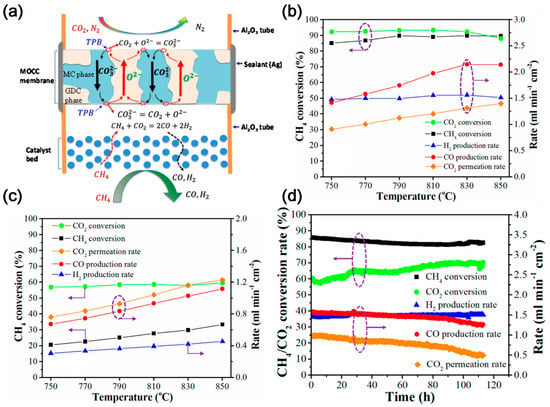
Figure 4. (a) Schematic illustration of a MOCC membrane reactor with a catalyst bed for electrochemical CO2 capture and catalytic DMR. TPB: triple phase boundary [31]. (b) Effect of temperature on the DMR performance of a GDC–MC membrane reactor with an NMP catalyst [31]. (c) Effect of temperature on the DMR performance of a GDC–MC membrane reactor with an NMP catalyst and an LNF catalyst [31]. (d) Stability of DOMR performance of the Ag–MC MECC membrane reactor at 800 °C with NMP catalyst. Feed gas: 75 mL min−1 N2, 15 mL min−1 CO2, and 10 mL min−1 O2; sweep gas: 0.94 mL min−1 CH4 and 50 mL min−1 Ar [32]. The dotted circles and arrows in the figure represent the axes corresponding to the curve.
The coupling of a dual-phase membrane reactor with a RWGS reaction was first reported by Chen et al. [34] to capture CO2 and produce CO. Under a sweep gas condition of 1% H2/He, the CO selectivity over LaNiO3 (LNO) catalyst is >96% at 550–750 °C. Additionally, La0.9Ce0.1NiO3-δ (LCNO) catalyst displays almost 100% CO selectivity and good thermal stability. The introduction of H2 during the purge test enhances the permeation of CO2. H2O in dual-phase membrane systems can reduce the activation energy, possibly due to the conduction of hydroxide ions through the membrane [35].
The dual-phase membrane reactor can also increase the H2 yield by removing CO2. The concept of CO2 removal was first applied to the steam reforming of methane reaction (SMR) to produce high concentration of H2 by Wu et al. [36]. The asymmetric BYS-SDC-MC membrane reactor can convert methane into hydrogen via SMR reaction while simultaneously removing CO2 to achieve a high concentration of hydrogen, with 84% CO2 recovery (Figure 5a). The results of the mathematical model (Figure 5b) demonstrate that CO2 recovery from SMR in the CO2 permeated membrane reactor exceeds 99% and a pure H2 gas stream without CO gas can be achieved [37].
Figure 5. (a) Schematic illustration of membrane reactor [37]. (b) Comparison of CO2 flux and recovery of the membrane reactor between experimental results and modeling results. Conditions: sweep: He = 200 mL min−1; feed: CH4 = 5 mL min−1; S/C = 3; both sides of the membrane were at 1 atm [37]. (c) Schematic illustration of (c1) Membrane reactor; (c2) Co-fed fixed bed reactor; (c3) Charge species transport and surface reactions in the membrane reactor; (c4) Reaction pathway diagram. 2D axial symmetric computational domain of (c5) Membrane reactor; (c6) Co-fed fixed bed reactor [38].
The dual-phase membrane reactors can also be utilized for the oxidative coupling of methane (OCM) reaction. The high oxygen partial pressure in conventional OCM reactors often results in low C2 selectivity. Li et al. [38] combined CO2/O2 transport membrane (CTM) and OCM reaction to build a new membrane reactor model (Figure 5c), which shows higher conversion of CH4 than the traditional fixed-bed reactor model and better coking resistance. Based on this, Zhang et al. [39] developed a membrane reactor combining SDC-NiO-MC and 2%Mn–5%Na2WO4/SiO2 catalyst. The co-captured CO2/O2 mixture converts CH4 into C2H6 over the 2%Mn–5%Na2WO4/SiO2 catalyst, followed by thermal cracking into C2H4 and H2. In the presence of CO2, the O2 partial pressure is reduced, thereby reducing the possibility of re-oxidation of C2 products, which leads to a higher C2 selectivity.
Table 2. Summary of electrochemical membrane materials integrated reactors.
| Membrane | Catalyst | Reaction | Ref. |
|---|---|---|---|
| La0.6Sr0.4Co0.8Fe0.2O3-δ Li-Na-K |
10 wt%Ni-/γ-Al2O3 | DMR | [29] |
| Ce0.8Gd0.2O1.9 Li-Na |
Ni-MgO-1 wt% Pt LaNi0.6Fe0.4O3-δ | DMR | [31] |
| Ag Li-Na |
Ni-MgO-1 wt% Pt | DOMR | [32] |
| NiO-SDC Li-Na |
Ni-MgO-1 wt% Pt | DOMR | [40] |
| Ce0.8Gd0.2O1.9 Li-Na |
5 wt% Cr2O3- ZSM-5 | Ethane-to-Ethylene | [33] |
| Ce0.9Pr0.1O2-δ-Pr0.6Sr0.4Fe0.5Co0.5O3-δ Li-Na-K |
10 wt%Ni-/γ-Al2O3 | DOMR | [41] |
| LNO/SDC Li-Na |
LNO/LCNO | RWGS | [34] |
| BYS-SDC Li-Na-K |
Ni-based catalyst (HiFUEL R110) | SMR | [36] |
| γ-LiAlO2-Ag Li-Na-K |
γ-LiAlO2-Ag | Syngas production | [42] |
| NiO-SDC Li-Na |
2%Mn-5%Na2WO4/SiO2 | OCM | [39] |
Note: Li-Na-K = Li2CO3: Na2CO3:K2CO3 with ratio of 42.5:32.5:25 mol%; Li-Na = Li2CO3: Na2CO3 with ratio of 52:48 mol%.
2.4. CO2 Capture and Conversion over Dual-Function Materials (DFMs)
Inorganic metal dual-functional materials (DFMs) combine the capture and conversion of CO2 into organic compounds. DFMs consist of the CO2 absorption component and the catalytic CO2 conversion component. The former is usually composed of alkali metal oxides or carbonates whilst the latter comprises metal-based catalysts, such as Ru, Rh, and Ni for DMR, dry ethane reforming (DER), RWGS reaction, and CO2 methanation [43,44,45]. Those processes can be well integrated with CO2 absorption considering their similar operating temperatures.
In 2018, Kim et al. [46] proposed and demonstrated the feasibility of using DFM to capture CO2 and convert it in situ with renewable CaO as an absorber of CO2 and Ni/MgO-Al2O3 as a catalyst for DMR. The concentration of CO2 in the exhaust gas is less than 0.08%, and the ratio of H2 to CO is 1.06. Tian et al. [47] synthesized CaO-Ni DFM using a sol–gel method. The ratio of H2 to CO is 1.1, which is similar to Kim’s results [46]. Methane decomposition can occur simultaneously during the reaction. The deposited carbon facilitates the reaction of CO2 with CaO-Ni; the CH4 conversion rate can reach 86%. Compared to other processes for DMR, the in situ consumption of CO2 on the catalyst surface promotes a positive shift of equilibrium in favor of CaCO3 decomposition, which allows the otherwise energy-intensive calcium cycling process to proceed at lower temperatures, thus further alleviating the deactivation problem during calcination. The energy consumed is 22% lower than the conventional consumption. In order to improve the activity and stability of Ni-CaO in the DMR reaction [48], CeO2 was added to the support, with a Ca:Ce molar ratio of 85:15. The resultant catalyst demonstrates good stability and maintains ≈ 80–90% activity over nine cycles at 650 °C, which is over two times better than the material without CeO2 modification (Figure 6a). The dispersion of Ni and the reducibility of NiO are improved, enhancing the DMR activity. Due to the high mobility of lattice oxygen in CeO2, the CeO2 modification can inhibit the accumulation of non-active carbon on Ni.
Figure 6. (a) Comparison of cyclic CO2 capture-release stability of the as-prepared bifunctional materials at 650 °C [48]. (b) CO2 sorption capacities and CO productivities of Ni/CS, Ni/CS–P30–C and Ni/CS–P30–C–P at 650 °C within 17 cycles [49] (The symbols indicate different preparation methods. The yellow and blue lines represent CO2 sorption capacities and CO productivities, respectively). Cyclic CO2 capture and conversion reactions of (c) Ca1Ni0.1; and (d) Ca1Ni0.1Ce0.033 [50].
In addition to DMR, the captured CO2 can be also converted into methane over DFM (Table 3). The Farrauto group is a pioneer in the study of CO2-Met DFM. They prepared DFM using Ru as the metal catalyst, CaO as the CO2 absorbent, and γ-Al2O3 as a carrier [51]. At 320 °C and 10% CO2/N2, CO2 capture was carried out followed by methane production reaction for 2 h with 4% H2/N2. The 5% Ru-10% CaO/γ-Al2O3 exhibits the highest activity. In the CO2 capture cycle test with presence of steam, the purity of obtained methane can reach up to 99%. Duyar et al. [52,53] synthesized Ru- and Rh-based DFM from the nitrates of Ru and Rh, respectively, via impregnation. The Rh-based DFMs show better activity. However, their high cost limits large-scale application. In order to reduce the cost, Bermejo-Lopez et al. studied DFMs using Ni as the catalyst [54,55]. Ni-CaO/γ-Al2O3 and Ni-Na2CO3/γ-Al2O3 with different Ni loadings were synthesized via impregnation. The methane yield of 10% Ni-Na2CO3/γ-Al2O3 is 186 μmol CH4 g−1 at 400 °C and it is 142 μmol CH4 g−1 at 520 °C for 15% Ni-CaO/γ-Al2O3. In addition, Al-Mamoori et al. [56] prepared DFMs using Ni-impregnated CaO- and MgO-based double salts as CO2 absorber and catalyst, respectively, and γ-Al2O3 as the carrier. The CO2 absorption was first saturated in a 10% CO2/N2 atmosphere at 650 °C and 1 bar, and then a 5% C2H6/N2 mixture was passed into the reactor to react with CO2. The Ni@(K-Ca)/γ-Al2O3 system exhibits the highest CO2 absorption and 100% C2H6 conversion. Nevertheless, Ni usually requires high reduction temperature and is very sensitive to O2 in the feed gas, and its long-term stability remains to be improved for the CO2-Met reaction.
Table 3. Carbon dioxide methanation dual-functional materials summary.
| DFM | Condition (°C) | Ref. | |
|---|---|---|---|
| Absorption | Reaction | ||
| Ni-CaO/Al2O3 | 280–520 10% CO2/Ar | 280–520 10% H2/Ar | [55] |
| Ni-Na2CO3/Al2O3 | 280–520 10% CO2/Ar | 280–520 10% H2/Ar | |
| Ni-Na2O/Al2O3 | 320 7.5% CO2/N2 and 7.5% CO2, 4.5% O2, 15% H2O/N2 | 320 15% H2/N2 | [52] |
| Ru-Na2O/γ-Al2O3 | 320 15% CO2/N2 | 320 20% H2/N2 | [57] |
| Rh-CaO/γ-Al2O3 | 320 10% CO2/N2 | 320 2% H2/N2 | |
| Ru-CaO/γ-Al2O3 | 280–400 1.4% CO2/Ar and 11% CO2/Ar | 280–400 10% H2/Ar | [54] |
| Ru-Na2CO3/γ-Al2O3 | 280–400 1.4% CO2/Ar and 11% CO2/A | 280–400 10% H2/Ar | |
| Ru-CaO/γ-Al2O3 | 320 10% CO2/air and 8% CO2/21% H2O/air | 320 5% H2/N2 | [51] |
| Ru-CaO/γ-Al2O3 | 320 10% CO2/N2 | 320 4% H2/N2 | [57] |
| Ru-CaO/γ-Al2O3 | 320 7.5% CO2, 4.5% O2, 15% H2O/N2 | 320 5% H2/N2 | [58] |
| Ru-Na2CO3/γ-Al2O3 | 320 7.5% CO2/N2 and 7.5% CO2, 4.5% O2, 15% H2O/N2 |
320 5% H2/N2 | [59] |
| Ru-Na2O/γ-Al2O3 | 250–350 7.5% CO2, 4.5% O2, 15% H2O/N2 | 250–350 15% H2/N2 | [60] |
DFMs can also convert the captured CO2 into valuable chemicals via RWGS reactions and ethane reforming. To investigate the effect of preparation methods on the performance of DFMs, Wang et al. [49] synthesized DFMs using three different methods, namely wet-mixing, acidification/impregnation, and acidification/impregnation combined with citric acid complexation. The Ni/CS-P30-C, prepared via acid pretreatment of carbide slag followed by citric acid complex, exhibits a high CO2 sorption capacity (13.28 mmol g−1 DFMs) and a great CO productivity (5.12 mmol g−1 DFMs) at 650 °C, and also demonstrates better CO2 capture and in situ conversion (Figure 6b). Sun et al. [50] investigated the effect of Ce loading onto the Ca-Ni-based DFM on the performance of CO2 capture and RWGS. The DFM with Ca:Ni:Ce = 1:0.1:0.033 (molar ratio) exhibits high cycling stability, and 100% CO selectivity (Figure 6c,d). The addition of CeO2 effectively prevents the growth and aggregation of NiO and CaO species.
The primary concerns are catalyst deactivation during the reaction, carbon deposition, and matching capture and conversion rates, along with the requirement of evaluating the life cycle and economics of the integrated system. Therefore, further research is necessary in the future on aspects, such as reaction atmosphere, temperature, life cycle, economic analysis, and industrial applications.
This entry is adapted from the peer-reviewed paper 10.3390/molecules28114500
This entry is offline, you can click here to edit this entry!