2. Long-Term Neurological Damage and the Role of Oxidative Stress
2.1. Neuroinvasive and Neurotoxic Potential of Coronavirus Linked to Neurodegeneration
The CNS has been proven to be susceptible to viral infections [
3,
4,
27,
28]. A series of recent studies have demonstrated the neuroinvasive potential of SARS-CoV-2, like the previously established neurovirulence of human coronaviruses, such as SARS-CoV, MERS-CoV, HcoV-229E, and HcoV-OC43 [
28,
29,
30,
31,
32]. According to Boroujeni et al., COVID-19 impacts the cerebral cortex of patients. This effect is characterized by activated microglia that amplify the inflammatory activation of astrocytes and is accompanied by low glutathione levels and upregulation of inflammation [
33]. Palpagama et al. asserted that the activation of glial cells is involved in the pathology of neurodegenerative disorders [
34]. The elevated number of such cells can be associated with neuroinflammation and brain tissue damage.
Other researchers have detected the presence of SARS-CoV-2 RNA in the cerebrospinal fluid after the virus infects the patient’s CNS and causes meningitis and encephalitis [
10,
32,
35,
36]. Alternatively, evidence has revealed that 50% of patients with COVID-19 develop intestinal inflammation [
37,
38,
39]. These results support the gut-driven inflammation hypothesis of Parkinson’s disease (PD) pathogenesis, which starts in the intestines and advances through inflammation toward the CNS. The associated increased levels of α-synuclein initiate aggregation in the gut and brain [
40]. Magnetic resonance imaging (MRI) data have also shown brain alteration in the cortical region (posterior gyrus rectus) that is associated with olfaction. This fact suggests that SARS-CoV-2 can invade the brain through the olfactory pathway and cause olfactory dysfunction, among other neurological disorders [
41]. Additionally, olfaction-related brain changes in the posterior gyrus rectus of the brain have been observed using magnetic resonance imaging (MRI) [
41]. This finding suggests that SARS-CoV-2 can enter the brain via the olfactory pathway and induce neurological diseases, such as olfactory impairment and other neurological conditions.
It is hypothesized that the neuroinvasive potential of SARS-CoV-2 plays a subordinate role in the pathogenesis of long COVID [
4,
42,
43,
44]. The virus enters the CNS and the PNS via the engagement of hematogenous or transsynaptic pathways (through the nasal cavity or the bloodstream) and triggers neuroinflammation [
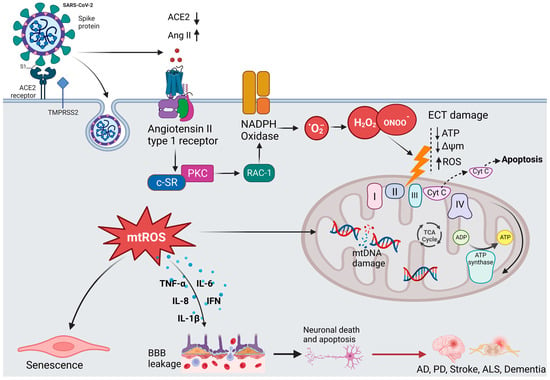
45]. As shown in
Figure 3, SARS-CoV-2 invades the host cells by binding to the ACE2 receptor with its spike (S) protein and then priming with the S protein through the activities of the transmembrane protease, serine 2 (TMPRSS2) [
46]. ACE2 is highly expressed in the brain of humans and animals, notably in some brain locations, such as the choroid plexus and paraventricular nuclei of the thalamus and in non-neuron cells (mainly astrocytes and oligodendrocytes) as well as various vessel calibers in the frontal cortex [
47]. The presence of ACE2 in primary human brain microvascular endothelial cells (hBMVECs) alleviates the ability of SARS-CoV-2 to compromise the activity of the BBB [
47,
48]. Coronavirus infections are primarily associated with cytokine production, inflammation, and apoptosis in accordance with the pathophysiological process of oxidative stress [
49].
Figure 3. The hypothesized mechanism of coronavirus-induced neuronal damage via oxidative stress and mitochondria dysfunction. SARS-CoV-2 infection occurs when TMPRSS2 primes the spike proteins for proteolysis, allowing the binding to ACE2. The affinity interaction triggers the binding of Ang II to the angiotensin type 1 receptor (AT1R), activating NADPH oxidase. This leads to mitochondrial electron transport chain (ETC) damage via the release of oxidative and nitrosative species, subsequently increasing the formation of mitochondrial reactive oxygen species (mtROS). The signaling pathways mediated by mtROS trigger the production of inflammatory cytokines, which can compromise the blood–brain barrier, thus resulting in neuronal damage. Additionally, mtROS cause nuclear and mitochondrial damage, which prolongs mitochondrial dysfunction and encourages inflammatory senescence.
2.2. Oxidative Stress and Redox Signaling, Players in SARS-CoV-2 Neurological Damage
It has been shown that Parkinson’s disease and SARS-CoV-2 infection cause similar oxidative stress and the activation of nuclear factor kappa B (NF-κB) [
18,
50,
51]. In addition, activating pro-inflammatory mediators, such as IL-1 and IL-6, can lead to amyloid beta (Aβ) deposition and accumulation, thus establishing a link between neuroinflammation and Alzheimer’s disease [
14,
52,
53].
It has been demonstrated that the receptor-binding domain (RBD) of the SARS-CoV-2 spike 1 (S1) protein binds to Aβ and tau proteins and causes their aggregation [
54]. Additionally, SARS-CoV-2 infection has been linked to hypoxia and decreased oxygen levels [
53]. The mitochondria of brain cells may undergo a rise in anaerobic metabolism due to this process, which can increase the levels of lactic acid, lipid peroxides, and oxygen-free radicals and deplete the antioxidant system. Consequently, the BBB is compromised, which may result in CNS complications. [
36,
49].
In regard to the role of oxidative stress in chronic diseases, Aranda-Rivera et al. highlighted the importance of nuclear factor erythroid 2–related factor 2 (Nrf2), a ubiquitous protein (containing 605 amino acids) that can modulate cellular oxidative stress [
55]. During oxidative stress, Keap1 undergoes conformational change due to the oxidation of residues Cys 226, Cys 613, and Cys 624 by electrophiles and oxidants [
56]. This mechanism enables Nrf2 to escape ubiquitination, leading to its release into the nucleus and regulating the expression of a number of antioxidant and detoxifying enzymes, such as glutamate-cysteine ligase modifier (GCLM) subunits, heme oxygenase, NAD(P)H quinone dehydrogenase 1 (NQO1), glutathione S-transferase, and glutathione peroxidase [
57,
58].
It should also be emphasized that viral infections, such as SARS-CoV-2, have adverse effects on antioxidant systems and have been linked to phenomena involving the inhibition of Nrf2 and activation of the NF-kB pathway in favor of inflammation and oxidative stress. Olagnier et al. showed that during SARS-CoV-2 infection, the Nrf2 pathway is repressed, leading to the downregulation of heme oxygenase 1 (HO-1) and NQO1 [
59]. Therefore, several antioxidant enzymes that guard against oxidative stress, including glutathione peroxide, peroxiredoxin, thioredoxin reductase, and thioredoxin, are affected.
Numerous studies have shown that the phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway controls oxidative stress by activating the transcription factor FOXO3 and initiating the transcription of antioxidant proteins, such as SOD-2, peroxiredoxins (PRDXs) 3 and 5, which are found in the mitochondria, and catalase, which is found in the peroxisomes [
60,
61,
62]. Peroxisome-proliferator-activated receptor coactivator 1 (PGC-1), the master biogenesis regulator that promotes the transcription of antioxidant enzymes, interacts with the FOXO3 transcription factor to control oxidative stress in the mitochondria [
61]. However, research has revealed that the ability of PGC-1 to promote gluconeogenesis and fatty acid oxidation is inhibited by protein kinase Akt2/protein kinase B (PKB), which acts as an intermediary trigger of phosphorylation and inhibition [
62].
NF-κB is another transcription factor that regulates stress responses. It is activated by the phosphorylation of I-κB, thanks to the I-κB kinase (IKK) complex. In vitro studies by Wu et al. have reported that sustained exposure of human lens epithelial cells (HLECs) to increasing doses of H
2O
2 (50–100 μM) for 4 h attenuates the TNFα-induced degradation of I-κB, accompanied by the activation of NF-κB and proteasome activity by 50–80% [
63]. The obtained data have also indicated that the activation of NF-κB is an essential phenomenon that enables cells to recover from oxidative stress.
2.3. In Vivo and In Vitro Models of Oxidative Stress
Over the years, various oxidative stress models have been developed to study the pathogenesis of neurodegenerative disorders and discover new strategies for developing therapeutic agents. Such experimental techniques include the use of lipid peroxidation products, endogenous antioxidant depletion, mitochondrial respiratory chain inhibitors, neurotoxic agents (e.g., rotenone and
N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)), and ischemic brain damage models [
51]. The proposed in vitro cellular models and in vivo animal models have shed light on the molecular mechanisms underlying oxidative stress responses in the nervous system, such as cell survival and cell death. Among neurotoxic chemical agents, 6-hydroxydopamine (6-OHDA) has been used to induce neurotoxicity in the dopaminergic nigrostriatal system by inhibiting the mitochondrial electron transport chain of complexes I and IV and accelerating neuronal degeneration [
64,
65,
66].
6-OHDA has been regarded as an endogenous toxic factor in the pathogenesis of PD. The neurotoxin 6-OHDA induces excessive production and accumulation of ROS and, therefore, oxidative stress. In fact, 6-OHDA induces caspase-3 activation in the cells mediated by the Fas or mitochondrial pathways [
67]. Indeed, it has been demonstrated that MTPT/6-OHDA-induced NF-κB activation in SH-SY5Y neuroblastoma cells triggers caspase-3 activation, which results in the death of DCNs via the NF-κB pathway [
68,
69,
70].
In vitro studies have evaluated the toxic effects of 6-OHDA in dopaminergic (DArgic) cell cultures. For instance, Vestuto et al. used human neuroblastoma SH-SY5Y cells to assess the neuroprotective effect of cocoa extract (purified fractions) in a 6-OHDA-induced PD cellular model [
71]. Similarly, Chansiw et al. reported the protective effect of 1-(
N-acetyl-6-aminohexyl)-3-hydroxy-2-methyl pyridine-4-one (CM1) coupled with green tea extract (GTE) on iron-induced oxidative stress in SH-SY5Y cells [
72]. In a separate study, Chen et al. evaluated the protective effect of EGCG against 6-OHDA-induced neurotoxicity by using N27 dopaminergic neurons [
73].
The advantages of cellular systems for studying oxidative stress are their low cost, adaptability, modularity, reproducibility, compatibility with high-throughput screening, and interest in cell mechanical investigations without systemic interferences. Other studies have demonstrated that hypoxia can increase cells’ susceptibility to oxidative stress. The hypoxia-reoxygenation model is a relevant in vitro model of oxidative stress, given that cellular hypoxia appears to be a crucial signal that activates transcriptional regulators, specifically hypoxia-inducible factor-1 (HIF-1) [
74], nuclear factor kappa B (NF-κB) [
75], activator protein 1 (AP-1) [
76,
77], and some mitogen-activated protein kinase (MAPK) signaling pathways, and induces cell death and necrosis [
76,
77].
Genetically derived models of neurodegenerative diseases are gaining considerable interest because they are excellent surrogates, providing intrinsic validity to genetically based models of degenerative disorders [
78]. Scientific investigations have reported that knockout of genes, including PINK1, DJ-1, LRRK2, and LRRK1, in rats leads to age-dependent neurodegeneration of the dopaminergic neurons of PD [
78,
79,
80]. Moreover, mutation in the copper-zinc superoxide dismutase 1 (SOD1) gene has been associated with ALS, while the alteration of MAPT genes or progranulin is linked to frontotemporal dementia (FTD) [
81]. Other studies have highlighted that mutations in amyloid precursor protein (APP), presenilin 1, and presenilin 2 (PSEN1/2) are the main causes of autosomal dominant early-onset AD [
82,
83].
It has been documented that invertebrates can also mimic endogenous-generated ROS. Such a model has been widely implemented in Drosophila melanogaster and
Caenorhabditis elegans (
C. elegans) [
84,
85]. The initial longevity mutant, known as age-1, which contains increased levels of both SOD1 and catalase, is arguably the best-studied mutant in the nematode
C. elegans. The age-1 mutant exhibits more significant levels of both SOD1 and catalase enzymes because this gene, which encodes for phosphatidylinositol-3 kinase, confers a longer lifetime phenotype when silenced, along with improved resilience to several forms of stress [
85,
86,
87].
3. Ginkgo Biloba Extract (EGb) for Neuroprotection and Potential Regeneration from Long COVID Syndrome
3.1. Ginkgo Biloba Antioxidative and Anti-Inflammatory Effects
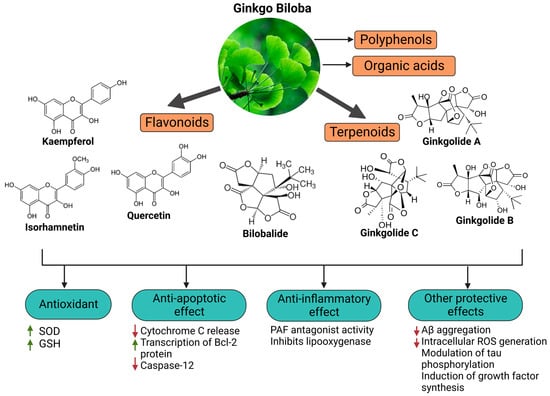
Ginkgo biloba (GB) is one of the medicinal plants that ameliorate capillary blood circulation, provide brain oxygenation, and thus improve age-related disorders. The ginkgo tree is monotypic and belongs to the class Ginkgoopsida, considered the oldest tree alive in the world (ginkgo species are from the Permian period, around 286–248 million years ago) [
23]. The currently available herbal medicines based on Ginkgo biloba extract (EGb) are Tebonin
® and Tanakan
®, which are mostly standardized on ginkgo flavone glycosides and terpene lactones—EGb 761
® [
26]. The standardized extract of Ginkgo biloba leaves includes 6% terpenoids, of which 3.1% are ginkgolides A, B, C, and J and 2.9% are bilobalide. It contains 24% flavonoid glycosides, including quercetin, kaempferol, and isorhamnetin, and 5–10% organic acids (
Figure 2) [
100]. Ginkgo biloba leaf extract is mentioned in the
British Herbal Compendium as a treatment for mild-to-severe dementia, including Alzheimer’s disease, and for the treatment of neurological symptoms attributed to loss of concentration and poor memory, confusion, depression, anxiety, vertigo, tinnitus, and headache [
101].
The neuroprotective effect of EGb 761 extract has been examined in a rat model of cerebral injury following ischemia/reperfusion (I/R). The treatment resulted in a decrease in MDA levels, the downregulation of pro-inflammatory cytokines (TNF-α and IL-1β), and an increase in the expression of anti-inflammatory cytokines (IL-10) and enzymatic SOD and myeloperoxidase (MPO) activities, which can control neurological impairments [
102]. It can be suggested that the beneficial effects of EGb on cerebral ischemia/reperfusion I/R injury result from the reduction in oxidative stress due to the inhibition of nitric oxide production and inflammation induced by I/R [
102].
Kaempferol is one of the most important constituents of Ginkgo biloba, and its action accounts for the upregulation of the glutamate-cysteine ligase catalytic (GCLC) subunit, brain-derived neurotrophic factor (BDNF), B-cell lymphoma protein 2 (Bcl-2), and GSH [
103,
104,
105]. Kaempferol inhibits ROS generation by scavenging free radicals and efficiently protects neuronal cells from oxidative injury. Additionally, it inhibits the production of pro-apoptotic proteins, including Bax and caspase-3, and modulates the downregulation of the NF-κB pathway to exert anti-apoptotic effects [
106]. A study by Zhou et al. showed that kaempferol can inhibit mitochondrial membrane transition (mPTP) opening and suppresses the release of cytochrome C via GSK-3β inhibition [
107]. Kaempferol is also involved in the inhibition of serotonin breakdown by monoamine oxidase, reduces neurotoxicity induced by 3-nitropropionic acid (3-NP), and induces the upregulation of heme oxygenase 1 (HMOX-1) [
108].
Quercetin, bilobalide, and isorhamnetin are other essential compounds in Ginkgo biloba extracts. Bilobalide decreases the expression of reactive species induced through H
2O
2, thus inhibiting ER stress [
109]. It can also suppress pro-inflammatory activation, NF-κB, and COX-2 activities [
110]. Studies have reported the beneficial effects of bilobalide in the upregulation of c-myc and p53 proteins, inhibition of the degradation of membrane phospholipids, and increased cellular proliferation of neurons in the hippocampus [
109,
111,
112]. According to Wang et al., pretreatment with bilobalide substantially reduced COX-2, iNOS, and phosphorylated p65 in sepsis-induced CLP mouse models, while inducing I-kB activation in the lungs [
111]. Additionally, bilobalide reduced oxidative stress by increasing HO-1 expression in lung tissues and antioxidative enzyme genes, including catalase, MnSOD, CuZnSOD, and GPx-1 [
111]. Moreover, bilobalide and EGb50 can modulate the expression of TLR4, NF-B, and MyD88, preventing the onset of acute lung injury (ALI) [
111,
113,
114]. Ginkgo biloba components may therefore prevent the onset of ALI and the cytokine storm syndrome in COVID-19 by inhibiting pro-inflammatory signaling via the NF-κB and TLR4 signaling pathways. A study showed that the administration of EGb50 substantially lowers TNF- and IL-1 levels and prevents the related signal transduction through the p38 MAPK and NF-B p65 pathways in LPS-stimulated microglial cells [
115]. In a separate study, EGb50 demonstrated a potential anti-inflammatory action by suppressing NLRP3-inflammasome-induced microglial activation [
116]. For comparison, isorhamnetin has been linked with the inhibition of apoptosis and the suppression of DNA fragmentation [
117].
In an enzymatic inhibition assay, it was established that ginkgolide A can act as an irreversible inhibitor against SARS-CoV-2 papain-like protease (PLpro) at a nontoxic dose of 1.79 µM [
20]. Similarly, quercetin, the primary EGb flavonoid component, inhibits SARS-CoV-2 3-chymotrypsin-like protease (3CLpro) and PLpro, with a corresponding docking energy of 6.25–4.62 kcal/mol, preventing SARS-CoV-2 replication [
118].
In a recent study, Liu et al. compared the antioxidant capacity of various Ginkgo biloba extracts by evaluating the mechanisms of ginkgolides A (GA), B (GB), K (GK), and bilobalide (BB) against oxidative stress caused by transient focal cerebral ischemia [
119].
In vivo studies have been performed in a developed middle cerebral artery occlusion (MCAO) model of cerebral ischemic injury using male SD rats, followed by reperfusion and Ginkgo biloba treatments [
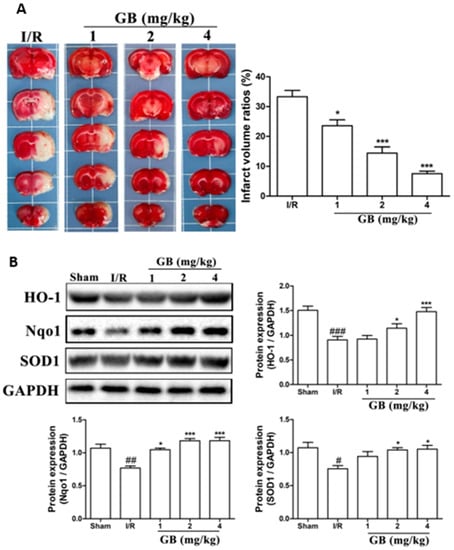
119]. Neuroblastoma cells (SH-SY5Y) were subjected to oxygen-glucose deprivation (OGD) for 4 h, followed by 6 h of reoxygenation using ginkgolides and bilobalide. The in vitro experimental findings revealed that GA, GB, GK, and BB significantly reduce ROS and increase SOD activities and protein levels, including HO-1 and Nqo1. Additionally, p-Akt and p-Nrf2 levels considerably increased following ginkgolide and BB treatments, with GB demonstrating greater efficacy than GA and GK. These upregulations could be reduced in a dose-dependent manner by LY294002, a PI3K inhibitor. The triphenyl tetrazolium chloride (TTC) staining performed demonstrated that GB significantly reduced the infarct volume ratios in MCAO rats in a dose-dependent manner (
Figure 4a). Additionally, GB markedly increased the amounts of the proteins HO-1, Nqo1, SOD, p-Akt, p-Nrf2, and Nrf2 through the modulation of the Akt/Nrf2 signaling pathway, shielding neurons from oxidative-stress-related damage (
Figure 4b) [
119].
Figure 4. Effects of Ginkgo biloba on infarct volume and expression of proteins associated with antioxidant effects in middle cerebral artery occlusion (MCAO) rat models. (A) MCAO rats were treated with various GB doses for 72 h, followed by triphenyl tetrazolium chloride (TTC) staining and statistical analysis of the cerebral infarct area. The results revealed a significant reduction in infarct volume ratios after GB treatments in a dose-dependent manner. (B) Results of Western blot analysis and semi-quantitative measurements of the levels of HO-1, Nqo1, and SOD1 proteins in the ischemic penumbra region of MCAO rats treated with various doses of Ginkgo biloba for 24 h. Antioxidant-related proteins in the MCAO group drastically decreased after cerebral ischemic injury compared to the normal group, while rats treated with various concentrations of GB showed a marked increase in the expression of HO-1, Nqo1, and SOD1. Data represented as the mean ± SD from eight rats of each group. (# p < 0.05, ## p < 0.01, and ### p < 0.001 vs. the sham group; * p < 0.05 and *** p < 0.001 vs. the I/R group).
3.2. Neuroprotective, Anti-Apoptotic, and Anxiolytic Drug Effects of EGb
Results from microarray experiments have established the neuroprotective effect of Egb 761 against ischemic-induced neuronal injury [
139]. The data revealed that the upregulation of Bcl-2 protein may be mediated by the activation of cAMP-response-element-binding protein (CREB) [
139]. EGb 761 increases CREB phosphorylation via the activation of PI3K/Akt and extracellular-signal-regulated kinase (ERK) signaling pathways [
140]. The consequently released BNDF protects the neurons against ischemia. Moreover, Tchantchou et al. affirmed that EGb 761 can reduce Aβ oligomerization and promote neurogenesis by the phosphorylation of CREB. This was evidenced by enhanced cell proliferation in the hippocampus of TgAPP/PS1 mice [
141]. Overall, these studies have demonstrated that flavonoids, the primary active constituents of EGb 761, may upregulate the CREB–BDNF pathway and therefore exert neuroprotection.
Combination therapy of EGb 761 with bone-marrow-derived mesenchymal stem cells (BMSCs) has shown a synergistic effect in animals with autoimmune encephalomyelitis. The therapeutic mechanism involves the inhibition of pro-inflammatory cytokines, demyelination, and protection axons and neurons [
142]. Other studies have documented that GA prevents p-Tau deposition and thus protects cells from toxicity associated with Tau hyperphosphorylation. Interestingly, the degree of dementia is closely correlated with the production of hyperphosphorylated Tau aggregates, making EGb 761 crucial to counteract the neurodegenerative process [
143].
Ginkgo biloba extracts, mainly flavonoids and ginkgolides, exert inhibitory effects on acetylcholinesterase activity. In fact, cholinergic agonists can reduce inflammation by blocking inflammatory signals, particularly the ubiquitous nuclear protein HMGB1 that is released by dying cells or activated innate immunity cells to promote inflammation [
144]. It has been suggested that nicotinic receptors nAChRs may control the expression of ACE2 and serve as a binding receptor for S1 protein, leading to an inflammatory response. However, EGb may counteract the central inhibitory and anti-inflammatory effects of GABAergic neurons, leading to increased cortical neuronal activity and an increased risk of convulsion [
145,
146]. However, meta-analysis research has proven that there is no convulsion risk associated with the anxiolytic action of EGb, which is mediated through the regulation of GABAergic neurons [
147]. In patients with dementia, EGb 761
® has shown promise in alleviating comorbid neurosensory symptoms and improving memory deficits [
147].
In another study, EGb 761 showed neuroprotective effects against oxidative-stress-induced apoptotic cell death by inhibiting apoptosis in a p53-dependent pathway, preventing mitochondrial membrane damage, reducing the release of cytochrome C from the mitochondria, upregulating the anti-apoptotic protein Bcl-2, and inhibiting PARP cleavage [
148]. The anti-apoptotic effects are schematically presented in
Figure 5.
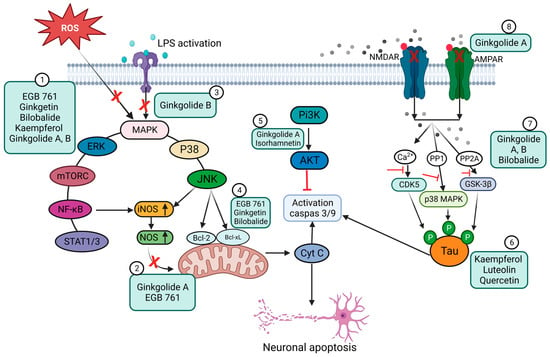
Figure 5. Schematic representation of the anti-apoptotic and anti-inflammatory effects of Ginkgo biloba extract (EGb) and its constituents. (1) Inhibition of ROS and suppression of the expression of pro-inflammatory mediators (e.g., COX-2 and NO) and pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) via the NF-κB signaling pathway. EGb can also inhibit the STAT 1/3 pathway. (2) Blocking of iNOS expression through a reduction in NO levels. (3) Inhibition of LPS-induced inflammatory response. (4) Prevention of mitochondrial oxidative stress by promoting the expression of anti-apoptotic proteins. (5) Inhibition of TLR4-NF-κB signaling through the PI3K/Akt pathway. (6) Prevention of the intracellular accumulation of p-Tau and cellular protection from Tau-hyperphosphorylation-related toxicity. (7) Blocking signaling pathways that involve CDK5, p38 MAPK, and GSK-3β. (8) Inhibition of NMDA and AMPA receptors, preventing the phosphorylation of c-Jun N-terminal kinase (JNK).
The work of Wang et al. showed that EGb may be used to increase cerebral blood flow and cognitive function by co-administration with 75 mg of aspirin toward the treatment of vascular cognitive impairment of non-dementia [
149]. In a randomized, double-blind exploratory study, the authors demonstrated that administration of EGb (Symfona
® forte) at a dose of 120 mg/twice daily for at least 6 months may improve dual-task-related gait performance in patients with MCI [
150]. Moreover, extensive work by Kuo et al. found that GA exhibits a strong therapeutic promise, like memantine, for treating AD by blocking NMDA and AMPA receptors. GA also suppresses c-Jun N-terminal kinase (JNK) activation at different doses (1–200 μM) in Aβ-induced neuronal depolarization in mice (
Figure 5) [
151].
In parallel, Yu et al. demonstrated the neuroprotective effect of Ginkgo biloba dropping pills (GBDP) in the amelioration of PD [
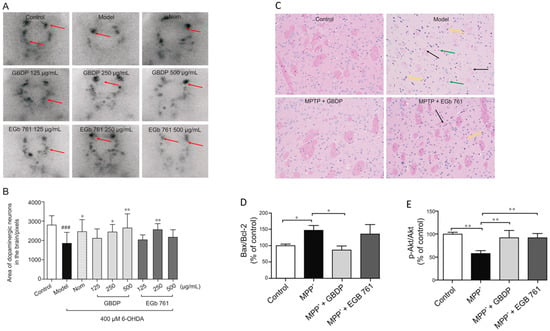
152]. In that study, the pharmacological effects of GBDP and EGb 761 were exploited in both in vivo and in vitro models of PD. Following GBDP and EGb 761 treatments, the viability of DA neurons in zebrafish was assessed via tyrosine hydroxylase immunostaining. Dopaminergic neurons in zebrafish were significantly lost after exposure to 400 mM 6-OHDA for 48 h. Nevertheless, administration of 250 or 500 mg/mL of GBDP or 250 mg/mL of EGb 761 rescued the death of dopaminergic neurons induced with 6-OHDA. No protection was observed with further GBDP and EGb 761 dosages (
Figure 6A,B). Moreover, GBDP reduced cognitive impairment and neuronal damage in MPTP-induced PD mice and reversed the effect of 6-OHDA-induced dopaminergic neuronal loss in zebrafish (
Figure 6C). In vitro findings revealed that the neuroprotective effects of GBDP can be mediated through the Akt/GSK3β pathway (
Figure 6D,E) [
152].
Figure 6. Representative images of dopaminergic neurons in the zebrafish brain, acquired using TH immunostaining. (A) Ginkgo biloba dropping pills (GBDP) prevented the loss of dopaminergic neurons induced by 6-OHDA. The red arrow shows dopaminergic neurons in the zebrafish brain. (B) The area of the dopaminergic neurons calculated for each group. ### p < 0.0001 vs. the control group; * p < 0.05 and ** p < 0.001 vs. the 6-OHDA group (n = 10 per group). (C) HE staining of brain sections of an MPTP-induced mouse model of Parkinson’s disease. The black arrow indicates significant diminished and loose nerve fiber components, which are lightly stained, while the yellow arrow shows intensive staining of the nuclei of several atrophied cells. Glial cells exhibited modest hyperplasia, as shown by the green arrow. (D,E) In MPP-treated human SH-SY5Y cells, GBDP administration reduced the Bax/Bcl-2 ratio and elevated Akt/GSK3β. ### p < 0.0001 vs. the MPP+ group.