Preeclampsia (PE) is a common obstetric disease characterized by hypertension, proteinuria, and multi-system dysfunction. It endangers both maternal and fetal health. Although hemostasis is critical for preventing bleeding complications during pregnancy, delivery, and post-partum, PE patients often develop a severe prothrombotic state, potentially resulting in life-threatening thrombosis and thromboembolism. The cause of this thrombotic complication is multi-factorial, involving endothelial cells, platelets, adhesive ligands, coagulation, and fibrinolysis. Increasing evidence has shown that hemostatic cells and factors undergo oxidative modifications during the systemic inflammation found in PE patients. However, it is largely unknown how these oxidative modifications of hemostasis contribute to development of the PE-associated prothrombotic state. This knowledge gap has significantly hindered the development of predictive markers, preventive measures, and therapeutic agents to protect women during pregnancy.
- Oxidative Stress
- Preeclampsia
- Prothrombotic State
Dear author, the following contents are excerpts from your papers. They are editable.
(Due to the lack of relevant professional knowledge, our editors cannot complete a complete entry by summarizing your paper, so if you are interested in this work. you may need to write some contents by yourself. A good entry will better present your ideas, research and results to other scholars. Readers will also be able to access your paper directly through entries.)
1. Introduction
Preeclampsia (PE) is a gestational disease that severely endangers maternal and fetal health and can develop into more severe complications (e.g., eclampsia) with long-term consequences. PE is defined as newly onset hypertension or proteinuria after 20 weeks of gestation without a prior history of hypertension, or newly onset hypertension with any of the following systemic manifestations: thrombocytopenia, renal insufficiency, impaired liver function, pulmonary edema, and severe headache without alternative diagnoses or visual impairments [1]. As the leading cause of maternal and perinatal morbidity and mortality, PE affects approximately 2% to 8% of pregnancies worldwide [2]. In an age-adjusted cohort study, the rate of PE in the US increased from 3.4% in 1980 to 3.8% in 2010 but the rate of severe PE increased by 322% during the same period [3]. This drastic increase was believed to be caused primarily by the age-cohort effect but it also highlights the need for improving pregnancy care. The current treatment of PE in clinical practice is delivery in time but antihypertension and spasmolysis may be used to control the progression of PE, prevent severe complications like eclampsia, and prolong the gestational week to improve maternal and fetal survival.
The maternal complications of PE include cerebrovascular diseases, acute renal failure, and subcapsular hematoma of the liver; the adverse perinatal outcomes include preterm delivery, fetal growth restriction (FGR), and fetal death [4]. PE patients also have a higher risk for long-term complications such as hypertension, ischemic heart disease, stroke, and venous thromboembolism [5]. Women with normal pregnancies often develop a systemic hypercoagulable state that progresses into a prothrombotic state in PE patients. In this review, we summarize the current knowledge of the role of oxidative stress in the development of this PE-associated prothrombotic state. This review focuses on linking oxidative stress to the prothrombotic state associated with PE. While both conditions develop frequently in patients with PE, they have been studied individually, without taking their causal relationship into consideration. Studying both conditions together will allow us to define this causal relationship, thus developing more accurate predictive markers and new targeted therapeutics for the patients.
Pathogenesis of PE
Placental villous lesions are found in 45.2% of PE patients compared with 14.6% of women with normal pregnancies, with vascular lesions being most common [14]. However, maternal conditions such as vascular disease, obesity, and autoimmune disease also contribute substantially to poor placentation and remodeling of the spiral arteries, leading to placental tissue ischemia and oxidative stress [15]. Placental tissue hypoxia is a hallmark event of PE, characterized by the overexpression of hypoxia-inducible transcription factor (HIF) [16] and poor angiogenesis in the placenta. It is associated with hypertension, proteinuria, and FGR [17].
The poor placental angiogenesis can result from dysregulation of growth factors such as vascular endothelial cell growth factor (VEGF) and the associated intracellular signaling pathways. For example, VEGF binds the fms-like tyrosine kinase receptor (Flt) to trigger proangiogenic signals. However, PE patients have significantly enhanced expression of soluble Flt 1 (sFlt-1) [18], an alternatively spliced variant of Flt that lacks the transmembrane and intracellular domains [19]. sFlt-1 binds VEGF and placental growth factor (PLGF) with a high affinity to competitively block VEGF binding to membrane-bound Flt and the resulting angiogenesis [19]. Placental biopsies have indeed shown that placentas from the majority of PE patients have poor trophoblastic invasion and vasculopathy of the spiral arteries that includes fibrin deposition, acute atherosis, and thrombosis [20]. More importantly, these local placental lesions can disseminate systemically by releasing soluble factors and extracellular vesicles into the maternal circulation [21,22], resulting in systemic endothelial injury and a prothrombotic state, as seen in PE patients.
There are two forms of PE: early-onset PE develops prior to 34 weeks of gestation and late-onset PE occurs after 34 gestational weeks. While both forms of PE are caused by syncytiotrophoblast stress, early-onset PE is closely associated with insufficient remodeling of spiral arteries and the resulting poor placentation, whereas late-onset PE often results from the restriction of placenta growth [15]. Early-onset PE is associated with more fetal death (adjusted odds ratios (OR) 5.8; 95% CI 4.0–8.3 vs. adjusted OR 1.3; 95% CI 0.8–2.0) and perinatal death/morbidity (adjusted OR 16.4; 95% CI 14.5–18.6 vs. adjusted OR 2.0; 95% CI 1.8–2.3) than late-onset PE [23].
2. Antioxidant Therapies in PE
2.1. Endogenous Antioxidants
The primary defense against oxidative stress are endogenous antioxidants such as SOD, catalase, and glutathione peroxidase that neutralize ROS. The level of SOD in the normal placental villous tissues increases from 8 weeks to 20 weeks of gestation [147], presumably to enhance antioxidant defense. The synthesis and enzymatic activity of all three enzymes are lower in placentas from PE patients than those from women with normotensive pregnancies [148,149,150]. Both SOD and catalase are also lower in the blood of PE patients than in that of normotensive pregnant women [151]. The rate-limiting enzyme heme oxygenase-1 (HO-1) catalyzes the potent oxidant heme into carbon monoxide, biliverdin, and free iron, thus serving as an antioxidant against cellular stresses like hypoxia and inflammation. The expression of HO-1 is reduced in placentas from PE patients [152,153]. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription activator that is expressed primarily in cytotrophoblast cells and regulates the expression of antioxidant proteins in these cells. Its expression is upregulated in the placentas of PE patients compared with gestation-matched normotensive subjects [154]. The chemical element selenium is an essential component of the antioxidants glutathione peroxidase and thioredoxin reductase. A meta-analysis by Xu et al. [155] reported that patients with PE have lower levels of plasma selenium than healthy pregnant women.
2.2. Therapeutic Antioxidants
Nanoscale selenium (Nano-Se) has been shown to ameliorate hyper-homocysteinemia-induced endothelial injury by inhibiting mitochondrial oxidative damage and apoptosis [156]. Selenium could also protect trophoblast cells from mitochondrial oxidative stress and ROS-mediated apoptosis [157,158], reducing the rate of PE [155]. Women who received 200 mg daily of CoQ10, a proton transfer agent in the mitochondrial electron transport chain, between 20 weeks of gestation and delivery, reduced their risk of PE compared with placebo controls [159]. CoQ10 has also been shown to improve specifically endothelial function [160]. Ergothioneine reduces the risk of PE [161], primarily by mitigating iron-induced oxidative stress. In a rat model of PE, ergothioneine ameliorated hypertension, reduced mitochondrial-specific H2O2 in the kidneys, and increased the fetal weight [162].
Both antiplatelet aspirin and anticoagulant heparin have been increasingly recommended for women at high risk of preeclampsia [163]. Apart from its conventional antithrombotic activity, aspirin has a modest ability of scavenging superoxide and it also contributes to the release of NO from the endothelium, which might result from the direct acetylation of eNOS [164]. Aspirin-triggered lipoxins inhibit the NADPH oxidase-mediated generation of ROS and nitrotyrosine in the endothelial cells [165,166]. Heparin has also been reported to reduce the plasma level of peroxides and increase the activity of SOD and catalase in red blood cells [167]. Low molecular weight heparin is shown to protect endothelial cells exposed to H2O2 [168]. However, it is not known whether aspirin and heparin reduce oxidative stress directly or by improving the systemic inflammation and tissue ischemia associated with the prothrombotic state of PE patients. Both the antioxidants pyrrolidine dithiocarbamate and N-acetylcysteine reduce the procoagulant activity of TNF-α-stimulated endothelial cells [72]. Antioxidants and NOX inhibitors significantly reduce platelet activation, aggregation, and thrombus formation [79,169].
3. Conclusions
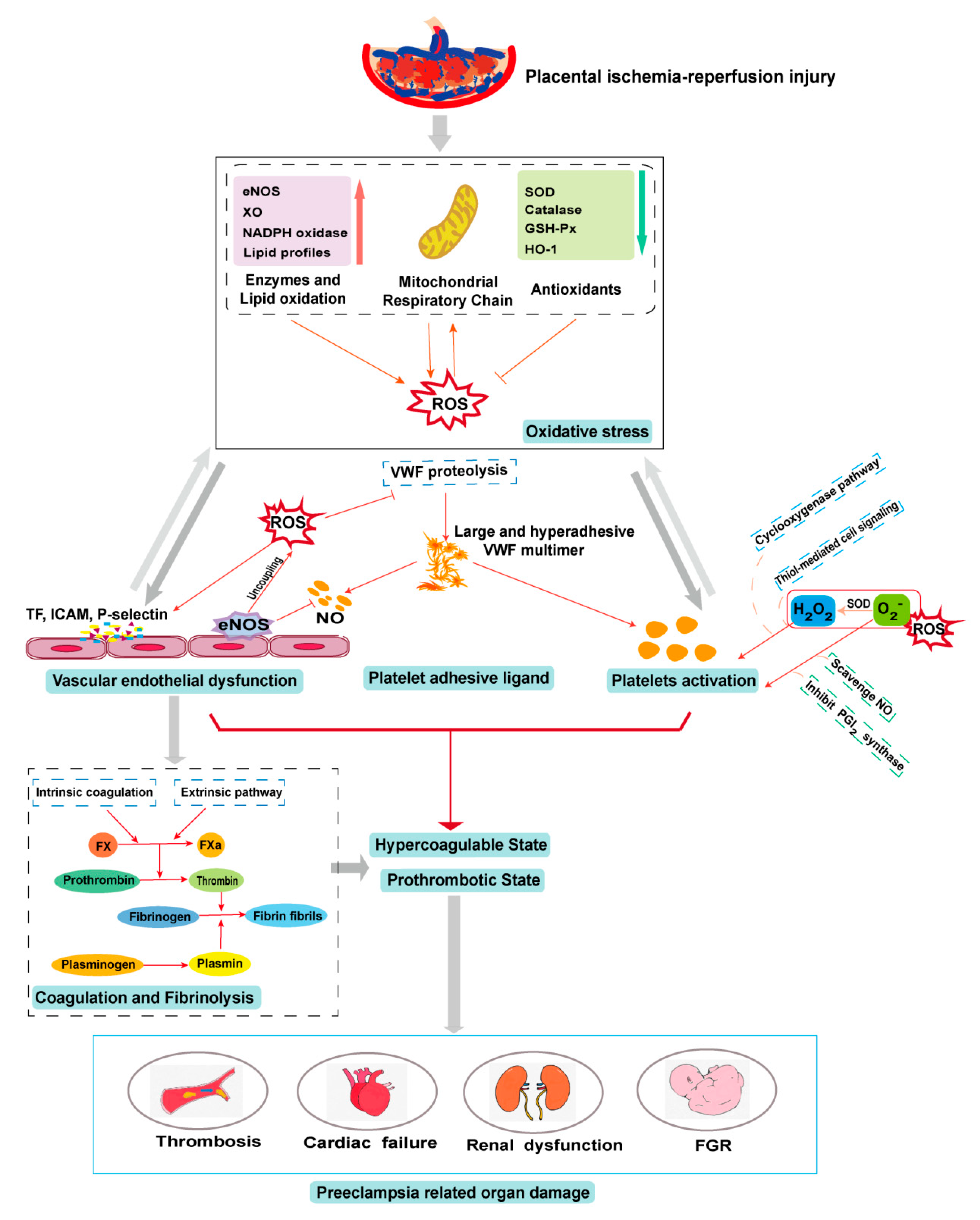
Oxidative stress plays a physiological role in placenta development but it can also cause placental pathologies and systemic conditions that initiate or propagate PE (Figure 2). Oxidative stress modifies the cells and molecules involved in all four components of hemostasis, resulting in hypercoagulable and prothrombotic states. These oxidative modifications are well documented for their biochemistry and cellular impacts but remain poorly understood for their specific roles in the pathogenesis of PE, especially regarding their impact on hemostasis in the condition of PE. Elucidating the role of oxidative stress in the pathogenesis of the PE-induced prothrombotic state could lead to new predictive markers and therapeutic targets for this severe pregnancy complication.
This entry is adapted from the peer-reviewed paper 10.3390/antiox9111139