Constantly evolving drug-resistant “superbugs” have caused an urgent demand for novel antimicrobial agents. Natural products and their analogs have been a prolific source of antimicrobial agents, even though a high rediscovery rate and less targeted research has made the field challenging in the pre-genomic era. With recent advancements in technology, natural product research is gaining new life. Genome mining has allowed for more targeted excavation of biosynthetic potential from natural sources that was previously overlooked. Researchers use bioinformatic algorithms to rapidly identify and predict antimicrobial candidates by studying the genome before even entering the lab.
- antimicrobial resistance
- genome mining
- natural products
- antibiotics
1. Introduction
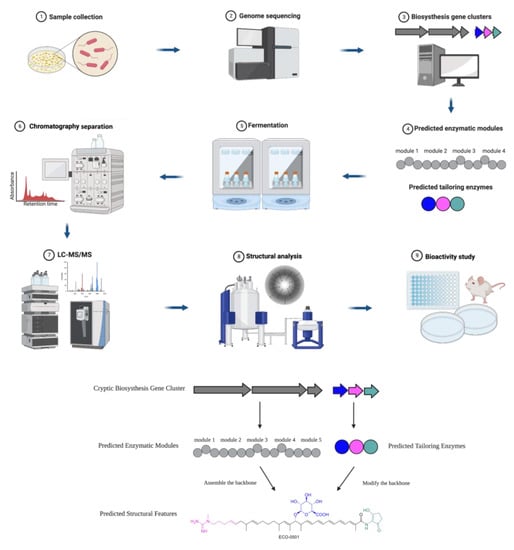
2. Genome Mining for Key Enzymes in Biosynthesis Gene Clusters
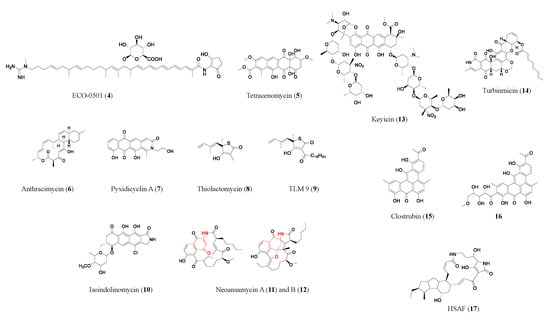
2.1. Polyketides
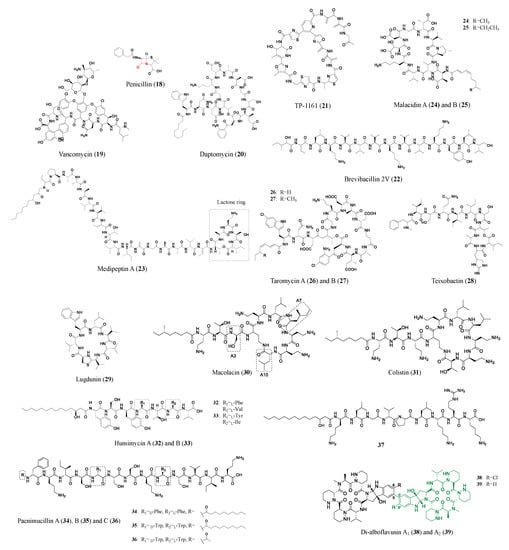
2.2. Nonribosomal Peptides
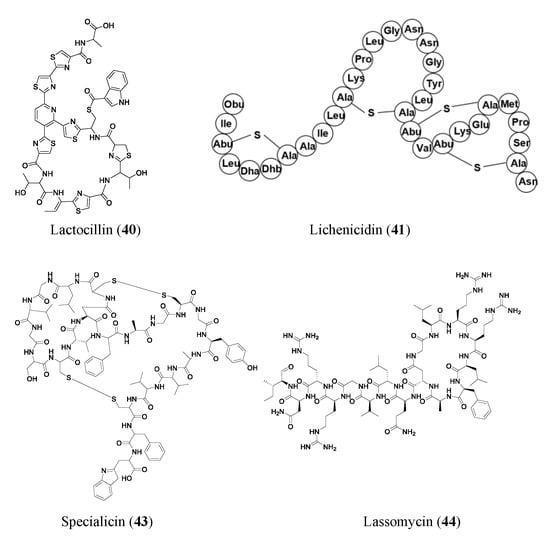
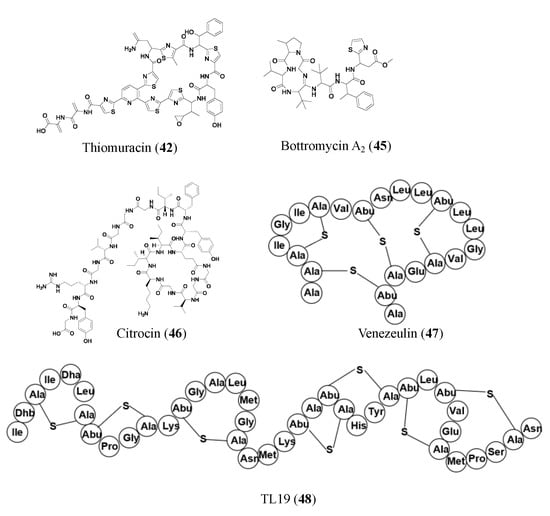
2.3. Ribosomally Synthesized and Post-Translationally Modified Peptides
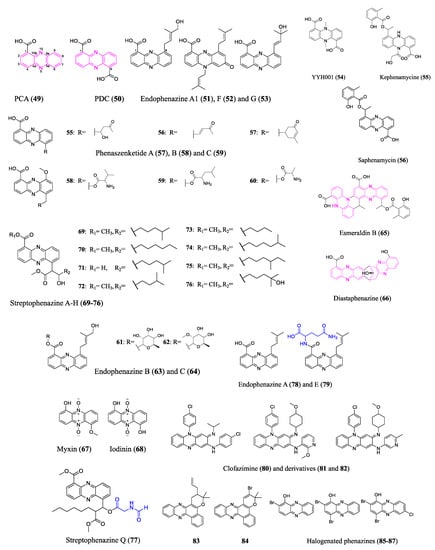
2.4. Phenazines
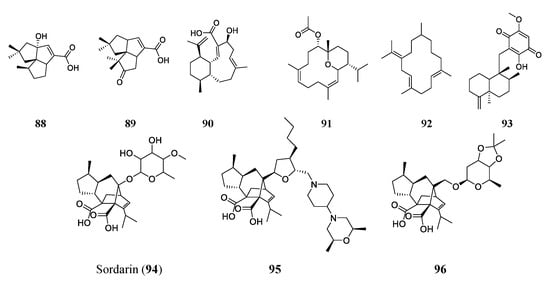
2.5. Terpenes/Terpenoids
This entry is adapted from the peer-reviewed paper 10.3390/molecules28104183
References
- Micarelli, I.; Paine, R.; Giostra, C.; Tafuri, M.A.; Profico, A.; Boggioni, M.; Di Vincenzo, F.; Massani, D.; Papini, A.; Manzi, G. Survival to amputation in pre-antibiotic era: A case study from a Longobard necropolis (6th-8th centuries AD). J. Anthropol. Sci. 2018, 96, 185–200.
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016.
- CDC. Antibiotic Resistance Threats in the United States 2019; Centers for Disease Control and Prevention, National Center for Emerging and Zoonotic Infectious Diseases, Division of Healthcare Quality Promotion, Antibiotic Resistance Coordination; Strategy Unit within the Division of Healthcare Quality Promotion, U.S. Department of Health and Human Services: Atlanta, GA, USA, 2019.
- CDC. COVID-19: U.S. Impact on Antimicrobial Resistance, Special Report 2022; Centers for Disease Control and Prevention, National Center for Emerging and Zoonotic Infectious Diseases, Division of Healthcare Quality Promotion, U.S. Department of Health and Human Services: Atlanta, GA, USA, 2022.
- Clardy, J.; Fischbach, M.A.; Walsh, C.T. New antibiotics from bacterial natural products. Nat. Biotechnol. 2006, 24, 1541–1550.
- Li, M.H.; Ung, P.M.; Zajkowski, J.; Garneau-Tsodikova, S.; Sherman, D.H. Automated genome mining for natural products. BMC Bioinform. 2009, 10, 185.
- Albarano, L.; Esposito, R.; Ruocco, N.; Costantini, M. Genome mining as new challenge in natural products discovery. Mar. Drugs 2020, 18, 199.
- Baltz, R.H. Genome mining for drug discovery: Progress at the front end. J. Ind. Microbiol. Biotechnol. 2021, 48, kuab044.
- Banskota, A.H.; McAlpine, J.B.; Sørensen, D.; Ibrahim, A.; Aouidate, M.; Piraee, M.; Alarco, A.-M.; Farnet, C.M.; Zazopoulos, E. Genomic analyses lead to novel secondary metabolites. J. Antiobiot. 2006, 59, 533–542.
- Shen, B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr. Opin. Chem. Biol. 2003, 7, 285–295.
- Helfrich, E.J.N.; Reiter, S.; Piel, J. Recent advances in genome-based polyketide discovery. Curr. Opin. Biotechnol. 2014, 29, 107–115.
- Bessa, L.J.; Buttachon, S.; Dethoup, T.; Martins, R.; Vasconcelos, V.; Kijjoa, A.; Martins da Costa, P. Neofiscalin A and fiscalin C are potential novel indole alkaloid alternatives for the treatment of multidrug-resistant Gram-positive bacterial infections. FEMS Microbiol. Lett. 2016, 363, fnw150.
- Hensler, M.E.; Jang, K.H.; Thienphrapa, W.; Vuong, L.; Tran, D.N.; Soubih, E.; Lin, L.; Haste, N.M.; Cunningham, M.L.; Kwan, B.P.; et al. Anthracimycin activity against contemporary methicillin-resistant Staphylococcus aureus. J. Antibiot. 2014, 67, 549–553.
- Panter, F.; Krug, D.; Baumann, S.; Müller, R. Self-resistance guided genome mining uncovers new topoisomerase inhibitors from myxobacteria. Chem. Sci. 2018, 9, 4898–4908.
- Tang, X.; Li, J.; Millan-Aguinaga, N.; Zhang, J.J.; O’Neill, E.C.; Ugalde, J.A.; Jensen, P.R.; Mantovani, S.M.; Moore, B.S. Identification of thiotetronic acid antibiotic biosynthetic pathways by target-directed genome mining. ACS Chem. Biol. 2015, 10, 2841–2849.
- Thong, W.L.; Shin-ya, K.; Nishiyama, M.; Kuzuyama, T. Discovery of an antibacterial isoindolinone-containing tetracyclic polyketide by cryptic gene activation and characterization of its biosynthetic gene cluster. ACS Chem. Biol. 2018, 13, 2615–2622.
- Fuoco, D. Classification framework and chemical biology of tetracycline-structure-based drugs. Antibiotics 2012, 1, 1–13.
- Wang, H.X.; Chen, Y.Y.; Ge, L.; Fang, T.T.; Meng, J.; Liu, Z.; Fang, X.Y.; Ni, S.; Lin, C.; Wu, Y.Y.; et al. PCR screening reveals considerable unexploited biosynthetic potential of ansamycins and a mysterious family of AHBA-containing natural products in actinomycetes. J. Appl. Microbiol. 2013, 115, 77–85.
- Li, S.; Li, Y.; Lu, C.; Zhang, J.; Zhu, J.; Wang, H.; Shen, Y. Activating a cryptic ansamycin biosynthetic gene cluster to produce three new naphthalenic octaketide ansamycins with n-pentyl and n-butyl side chains. Org. Lett. 2015, 17, 3706–3709.
- Liu, M.; Lu, C.; Tang, R.; Li, S.; Wang, H.; Shen, Y. Neoansamycins from Streptomyces sp. LZ35. RSC Adv. 2017, 7, 35460–35465.
- Adnani, N.; Chevrette, M.G.; Adibhatla, S.N.; Zhang, F.; Yu, Q.; Braun, D.R.; Nelson, J.; Simpkins, S.W.; McDonald, B.R.; Myers, C.L.; et al. Coculture of marine invertebrate-associated bacteria and interdisciplinary technologies enable biosynthesis and discovery of a new antibiotic, Keyicin. ACS Chem. Biol. 2017, 12, 3093–3102.
- Chanana, S.; Thomas, C.S.; Zhang, F.; Rajski, S.R.; Bugni, T.S. hcapca: Automated hierarchical clustering and principal component analysis of large metabolomic datasets in R. Metabolites 2020, 10, 297.
- Brkljača, R.; Urban, S. Recent advancements in HPLC-NMR and applications for natural product profiling and identification. J. Liq. Chromatogr. Relat. Technol. 2011, 34, 1063–1076.
- Danelius, E.; Halaby, S.; van der Donk, W.A.; Gonen, T. MicroED in natural product and small molecule research. Nat. Prod. Rep. 2021, 38, 423–431.
- Hansen, P.E. NMR of natural products as potential drugs. Molecules 2021, 26, 3763.
- Kim, L.J.; Ohashi, M.; Zhang, Z.; Tan, D.; Asay, M.; Cascio, D.; Rodriguez, J.A.; Tang, Y.; Nelson, H.M. Prospecting for natural products by genome mining and microcrystal electron diffraction. Nat. Chem. Biol. 2021, 17, 872–877.
- Zhang, F.; Zhao, M.; Braun, D.R.; Ericksen, S.S.; Piotrowski, J.S.; Nelson, J.; Peng, J.; Ananiev, G.E.; Chanana, S.; Barns, K.; et al. A marine microbiome antifungal targets urgent-threat drug-resistant fungi. Science 2020, 370, 974–978.
- Pidot, S.; Ishida, K.; Cyrulies, M.; Hertweck, C. Discovery of clostrubin, an exceptional polyphenolic polyketide antibiotic from a strictly anaerobic bacterium. Angew. Chem. Int. Ed. Engl. 2014, 53, 7856–7859.
- Shabuer, G.; Ishida, K.; Pidot, S.J.; Roth, M.; Dahse, H.-M.; Hertweck, C. Plant pathogenic anaerobic bacteria use aromatic polyketides to access aerobic territory. Science 2015, 350, 670–674.
- Süssmuth, R.D.; Mainz, A. Nonribosomal Peptide Synthesis-Principles and Prospects. Angew. Chem. Int. Ed. Engl. 2017, 56, 3770–3821.
- Engelhardt, K.; Degnes, K.F.; Kemmler, M.; Bredholt, H.; Fjaervik, E.; Klinkenberg, G.; Sletta, H.; Ellingsen, T.E.; Zotchev, S.B. Production of a new thiopeptide antibiotic, TP-1161, by a marine Nocardiopsis species. Appl. Environ. Microbiol. 2010, 76, 4969–4976.
- Zhao, X.; Wang, X.; Shukla, R.; Kumar, R.; Weingarth, M.; Breukink, E.; Kuipers, O.P. Brevibacillin 2V, a novel antimicrobial lipopeptide with an exceptionally low hemolytic activity. Front. Microbiol. 2021, 12, 693725.
- Zhou, L.; de Jong, A.; Yi, Y.; Kuipers, O.P. Identification, isolation, and characterization of medipeptins, antimicrobial peptides from Pseudomonas mediterranea EDOX. Front. Microbiol. 2021, 12, 732771.
- Hover, B.M.; Kim, S.H.; Katz, M.; Charlop-Powers, Z.; Owen, J.G.; Ternei, M.A.; Maniko, J.; Estrela, A.B.; Molina, H.; Park, S.; et al. Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nat. Microbiol. 2018, 3, 415–422.
- Yamanaka, K.; Reynolds, K.A.; Kersten, R.D.; Ryan, K.S.; Gonzalez, D.J.; Nizet, V.; Dorrestein, P.C.; Moore, B.S. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc. Natl. Acad. Sci. USA 2014, 111, 1957–1962.
- Reynolds, K.A.; Luhavaya, H.; Li, J.; Dahesh, S.; Nizet, V.; Yamanaka, K.; Moore, B.S. Isolation and structure elucidation of lipopeptide antibiotic taromycin B from the activated taromycin biosynthetic gene cluster. J. Antibiot. (Tokyo) 2018, 71, 333–338.
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459.
- Zipperer, A.; Konnerth, M.C.; Laux, C.; Berscheid, A.; Janek, D.; Weidenmaier, C.; Burian, M.; Schilling, N.A.; Slavetinsky, C.; Marschal, M.; et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516.
- Wang, Z.; Koirala, B.; Hernandez, Y.; Zimmerman, M.; Park, S.; Perlin, D.S.; Brady, S.F. A naturally inspired antibiotic to target multidrug-resistant pathogens. Nature 2022, 601, 606–611.
- Chu, J.; Vila-Farres, X.; Inoyama, D.; Ternei, M.; Cohen, L.J.; Gordon, E.A.; Reddy, B.V.; Charlop-Powers, Z.; Zebroski, H.A.; Gallardo-Macias, R.; et al. Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biol. 2016, 12, 1004–1006.
- Vila-Farres, X.; Chu, J.; Ternei, M.A.; Lemetre, C.; Park, S.; Perlin, D.S.; Brady, S.F. An optimized synthetic-bioinformatic natural product antibiotic sterilizes multidrug-resistant Acinetobacter baumannii-infected wounds. mSphere 2018, 3, e00528-17.
- Vila-Farres, X.; Chu, J.; Inoyama, D.; Ternei, M.A.; Lemetre, C.; Cohen, L.J.; Cho, W.; Reddy, B.V.B.; Zebroski, H.A.; Freundlich, J.S.; et al. Antimicrobials inspired by nonribosomal peptide synthetase gene clusters. J. Am. Chem. Soc. 2017, 139, 1404–1407.
- Guo, Z.; Li, P.; Chen, G.; Li, C.; Cao, Z.; Zhang, Y.; Ren, J.; Xiang, H.; Lin, S.; Ju, J.; et al. Design and biosynthesis of dimeric alboflavusins with biaryl linkages via regiospecific C–C bond coupling. J. Am. Chem. Soc. 2018, 140, 18009–18015.
- Poorinmohammad, N.; Bagheban-Shemirani, R.; Hamedi, J. Genome mining for ribosomally synthesised and post-translationally modified peptides (RiPPs) reveals undiscovered bioactive potentials of actinobacteria. Antonie Van Leeuwenhoek 2019, 112, 1477–1499.
- Der Torossian Torres, M.; de la Fuente-Nunez, C. Reprogramming biological peptides to combat infectious diseases. Chem. Commun. 2019, 55, 15020–15032.
- Donia, M.S.; Cimermancic, P.; Schulze, C.J.; Wieland Brown, L.C.; Martin, J.; Mitreva, M.; Clardy, J.; Linington, R.G. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 2014, 158, 1402–1414.
- Dischinger, J.; Josten, M.; Szekat, C.; Sahl, H.G.; Bierbaum, G. Production of the novel two-peptide lantibiotic lichenicidin by Bacillus licheniformis DSM 13. PLoS ONE 2009, 4, e6788.
- Shenkarev, Z.O.; Finkina, E.I.; Nurmukhamedova, E.K.; Balandin, S.V.; Mineev, K.S.; Nadezhdin, K.D.; Yakimenko, Z.A.; Tagaev, A.A.; Temirov, Y.V.; Arseniev, A.S.; et al. Isolation, structure elucidation, and synergistic antibacterial activity of a novel two-component lantibiotic lichenicidin from Bacillus licheniformis VK21. Biochemistry 2010, 49, 6462–6472.
- Hudson, G.A.; Zhang, Z.; Tietz, J.I.; Mitchell, D.A.; van der Donk, W.A. In vitro biosynthesis of the core scaffold of the thiopeptide thiomuracin. J. Am. Chem. Soc. 2015, 137, 16012–16015.
- Kaweewan, I.; Hemmi, H.; Komaki, H.; Harada, S.; Kodani, S. Isolation and structure determination of a new lasso peptide specialicin based on genome mining. Bioorg. Med. Chem. 2018, 26, 6050–6055.
- Gavrish, E.; Clarissa, S.; Cao, S.; Kandror, O.; Spoering, A.; Peoples, A.; Ling, L.; Fetterman, A.; Hughes, D.; Bissell, A.; et al. Lassomycin, a ribosomally synthesized cyclic peptide, kills Mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem. Biol. 2014, 21, 509–518.
- Cheung-Lee, W.L.; Parry, M.E.; Jaramillo Cartagena, A.; Darst, S.A.; Link, A.J. Discovery and structure of the antimicrobial lasso peptide citrocin. J. Biol. Chem. 2019, 294, 6822–6830.
- Horbal, L.; Marques, F.; Nadmid, S.; Mendes, M.V.; Luzhetskyy, A. Secondary metabolites overproduction through transcriptional gene cluster refactoring. Metab. Eng. 2018, 49, 299–315.
- Shimamura, H.; Gouda, H.; Nagai, K.; Hirose, T.; Ichioka, M.; Furuya, Y.; Kobayashi, Y.; Hirono, S.; Sunazuka, T.; Ōmura, S. Structure determination and total synthesis of bottromycin A2: A potent antibiotic against MRSA and VRE. Angew. Chem. 2009, 121, 932–935.
- Goto, Y.; Li, B.; Claesen, J.; Shi, Y.; Bibb, M.J.; van der Donk, W.A. Discovery of unique lanthionine synthetases reveals new mechanistic and evolutionary insights. PLoS Biol. 2010, 8, e1000339.
- van Staden, A.D.P.; van Zyl, W.F.; Trindade, M.; Dicks, L.M.T.; Smith, C. Therapeutic application of lantibiotics and other lanthipeptides: Old and new findings. Appl. Environ. Microbiol. 2021, 87, e0018621.
- Zhao, X.; Yin, Z.; Breukink, E.; Moll, G.N.; Kuipers, O.P. An engineered double lipid II binding motifs-containing lantibiotic displays potent and selective antimicrobial activity against Enterococcus faecium. Antimicrob. Agents Chemother. 2020, 64, e02050-19.
- Jiang, J.; Guiza Beltran, D.; Schacht, A.; Wright, S.; Zhang, L.; Du, L. Functional and structural analysis of Phenazine O-Methyltransferase LaPhzM from Lysobacter antibioticus OH13 and one-pot enzymatic synthesis of the antibiotic myxin. ACS Chem. Biol. 2018, 13, 1003–1012.
- Lin, C.; Yang, D. DNA recognition by a novel bis-intercalator, potent anticancer drug XR5944. Curr. Top. Med. Chem. 2015, 15, 1385–1397.
- Guttenberger, N.; Blankenfeldt, W.; Breinbauer, R. Recent developments in the isolation, biological function, biosynthesis, and synthesis of phenazine natural products. Bioorg. Med. Chem. 2017, 25, 6149–6166.
- Heine, D.; Martin, K.; Hertweck, C. Genomics-guided discovery of endophenazines from Kitasatospora sp. HKI 714. J. Nat. Prod. 2014, 77, 1083–1087.
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35.
- Shi, Y.-M.; Brachmann, A.O.; Westphalen, M.A.; Neubacher, N.; Tobias, N.J.; Bode, H.B. Dual phenazine gene clusters enable diversification during biosynthesis. Nat. Chem. Biol. 2019, 15, 331–339.
- Giddens, S.R.; Bean, D.C. Investigations into the in vitro antimicrobial activity and mode of action of the phenazine antibiotic d-alanylgriseoluteic acid. Int. J. Antimicrob. Agents. 2007, 29, 93–97.
- Wu, C.; van Wezel, G.P.; Hae Choi, Y. Identification of novel endophenaside antibiotics produced by Kitasatospora sp. MBT66. J. Antibiot. 2015, 68, 445–452.
- Li, Y.; Han, L.; Rong, H.; Li, L.; Zhao, L.; Wu, L.; Xu, L.; Jiang, Y.; Huang, X. Diastaphenazine, a new dimeric phenazine from an endophytic Streptomyces diastaticus subsp. ardesiacus. J. Antibiot. 2015, 68, 210–212.
- Rui, Z.; Ye, M.; Wang, S.; Fujikawa, K.; Akerele, B.; Aung, M.; Floss, H.G.; Zhang, W.; Yu, T.W. Insights into a divergent phenazine biosynthetic pathway governed by a plasmid-born esmeraldin gene cluster. Chem. Biol. 2012, 19, 1116–1125.
- Zhao, Y.; Qian, G.; Ye, Y.; Wright, S.; Chen, H.; Shen, Y.; Liu, F.; Du, L. Heterocyclic aromatic N-Oxidation in the biosynthesis of phenazine antibiotics from Lysobacter antibioticus. Org. Lett. 2016, 18, 2495–2498.
- Mitova, M.I.; Lang, G.; Wiese, J.; Imhoff, J.F. Subinhibitory concentrations of antibiotics induce phenazine production in a marine Streptomyces sp. J. Nat. Prod. 2008, 71, 824–827.
- Gomez-Escribano, J.P.; Bibb, M.J. Heterologous expression of natural product biosynthetic gene clusters in Streptomyces coelicolor: From genome mining to manipulation of biosynthetic pathways. J. Ind. Microbiol. Technol. 2014, 41, 425–431.
- Saleh, O.; Flinspach, K.; Westrich, L.; Kulik, A.; Gust, B.; Fiedler, H.-P.; Heide, L. Mutational analysis of a phenazine biosynthetic gene cluster in Streptomyces anulatus 9663. Beilstein J. Org. Chem. 2012, 8, 501–513.
- Saleh, O.; Bonitz, T.; Flinspach, K.; Kulik, A.; Burkard, N.; Mühlenweg, A.; Vente, A.; Polnick, S.; Lämmerhofer, M.; Gust, B.; et al. Activation of a silent phenazine biosynthetic gene cluster reveals a novel natural product and a new resistance mechanism against phenazines. Med. Chem. Commun. 2012, 3, 1009–1019.
- Lechartier, B.; Cole, S.T. Mode of action of clofazimine and combination therapy with benzothiazinones against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 4457–4463.
- Zhang, D.; Lu, Y.; Liu, K.; Liu, B.; Wang, J.; Zhang, G.; Zhang, H.; Liu, Y.; Wang, B.; Zheng, M.; et al. Identification of less lipophilic riminophenazine derivatives for the treatment of drug-resistant Tuberculosis. J. Med. Chem. 2012, 55, 8409–8417.
- Zhang, D.; Liu, Y.; Zhang, C.; Zhang, H.; Wang, B.; Xu, J.; Fu, L.; Yin, D.; Cooper, C.B.; Ma, Z.; et al. Synthesis and biological evaluation of novel 2-Methoxypyridylamino-substituted riminophenazine derivatives as antituberculosis agents. Molecules 2014, 19, 4380–4394.
- Liu, B.; Liu, K.; Lu, Y.; Zhang, D.; Yang, T.; Li, X.; Ma, C.; Zheng, M.; Wang, B.; Zhang, G.; et al. Systematic evaluation of structure-activity relationships of the riminophenazine class and discovery of a C2 pyridylamino series for the treatment of multidrug-resistant Tuberculosis. Molecules 2012, 17, 4545–4559.
- Coelho, T.S.; Silva, R.S.F.; Pinto, A.V.; Pinto, M.C.F.R.; Scaini, C.J.; Moura, K.C.G.; Almeida da Silva, P. Activity of β-lapachone derivatives against rifampicin-susceptible and -resistant strains of Mycobacterium tuberculosis. Tuberculosis 2010, 90, 293–297.
- Borrero, N.V.; Bai, F.; Perez, C.; Duong, B.Q.; Rocca, J.R.; Jin, S.; Huigens Iii, R.W. Phenazine antibiotic inspired discovery of potent bromophenazine antibacterial agents against Staphylococcus aureus and Staphylococcus epidermidis. Org. Biomol. Chem. 2014, 12, 881–886.
- Garrison, A.T.; Abouelhassan, Y.; Norwood, V.M.I.V.; Kallifidas, D.; Bai, F.; Nguyen, M.T.; Rolfe, M.; Burch, G.M.; Jin, S.; Luesch, H.; et al. Structure–activity relationships of a diverse class of halogenated phenazines that targets persistent, antibiotic-tolerant bacterial biofilms and Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 3808–3825.
- Garrison, A.T.; Abouelhassan, Y.; Kallifidas, D.; Bai, F.; Ukhanova, M.; Mai, V.; Jin, S.; Luesch, H.; Huigens, R.W., 3rd. Halogenated phenazines that potently eradicate biofilms, MRSA persister cells in non-biofilm cultures, and Mycobacterium tuberculosis. Angew. Chem. Int. Ed. Engl. 2015, 54, 14819–14823.
- Huigens, R.W., 3rd; Abouelhassan, Y.; Yang, H. Phenazine antibiotic-inspired discovery of bacterial biofilm-eradicating agents. Chembiochem 2019, 20, 2885–2902.
- Reusch, W. Textbook of Organic Chemistry. Available online: https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Lipids/Properties_and_Classification_of_Lipids/Terpenes (accessed on 6 January 2023).
- Soderberg, T.; Reusch, W.; Farmer, S.; Kennepohl, D. Terpenoids. Available online: https://chem.libretexts.org/Courses/Athabasca_University/Chemistry_360%3A_Organic_Chemistry_II/Chapter_27%3A_Biomolecules_-_Lipids/27.05_Terpenoids (accessed on 6 January 2023).
- Li, H.; Li, H.; Chen, S.; Wu, W.; Sun, P. Isolation and identification of pentalenolactone analogs from Streptomyces sp. NRRL S-4. Molecules 2021, 26, 7377.
- Zhu, C.; Xu, B.; Adpressa, D.A.; Rudolf, J.D.; Loesgen, S. Discovery and biosynthesis of a structurally dynamic antibacterial diterpenoid. Angew Chem. Int. Ed. Engl. 2021, 60, 14163–14170.
- Al-Footy, K.O.; Alarif, W.M.; Asiri, F.; Aly, M.M.; Ayyad, S.-E.N. Rare pyrane-based cembranoids from the Red Sea soft coral Sarcophyton trocheliophorum as potential antimicrobial–antitumor agents. Med. Chem. Res. 2015, 24, 505–512.
- Kumar, R.; Subramani, R.; Aalbersberg, W. Three bioactive sesquiterpene quinones from the Fijian marine sponge of the genus Hippospongia. Nat. Prod. Res. 2013, 27, 1488–1491.
- Kudo, F.; Matsuura, Y.; Hayashi, T.; Fukushima, M.; Eguchi, T. Genome mining of the sordarin biosynthetic gene cluster from Sordaria araneosa Cain ATCC 36386: Characterization of cycloaraneosene synthase and GDP-6-deoxyaltrose transferase. J. Antibiot. 2016, 69, 541–548.
- Hanadate, T.; Tomishima, M.; Shiraishi, N.; Tanabe, D.; Morikawa, H.; Barrett, D.; Matsumoto, S.; Ohtomo, K.; Maki, K. FR290581, a novel sordarin derivative: Synthesis and antifungal activity. Bioorg. Med. Chem. Lett. 2009, 19, 1465–1468.
- Bueno, J.M.; Chicharro, J.; Fiandor, J.M.; Gómez de las Heras, F.; Huss, S. Antifungal sordarins. Part 4: Synthesis and structure–activity relationships of 3′,4′-fused alkyl-tetrahydrofuran derivatives. Bioorg. Med. Chem. Lett. 2002, 12, 1697–1700.
- Bommineni, G.R.; Kapilashrami, K.; Cummings, J.E.; Lu, Y.; Knudson, S.E.; Gu, C.; Walker, S.G.; Slayden, R.A.; Tonge, P.J. Thiolactomycin-based inhibitors of bacterial β-ketoacyl-ACP synthases with in vivo activity. J. Med. Chem. 2016, 59, 5377–5390.