In the early 2000s, technological advances in genome sequencing and bioinformatics on filamentous fungi began to reveal a discrepancy between the number of biosynthetic gene clusters (BGCs) encoding the biosynthesis of fungal secondary metabolites and the actual number of identified fungal compounds from the target strain. The discovery of cryptic BGCs in microorganisms, including fungi, has spurred the development of new experimental methodologies for identifying the secondary metabolites of these clusters, which led to the realization that they have the potential to produce novel specialized metabolites, giving rise to a new field of research called genome-guided natural product discovery.
1. Introduction
Natural products have been recognized as crucial sources for new drug discovery. Over the past 38 years, more than half of the clinical drugs that have been approved by the FDA were derived from natural sources, and natural products still hold promising potential for discovering novel drug candidates and bioactive chemical templates [
1,
2]. Fungi, in particular, offer an incredibly prolific and diverse array of bioactive secondary metabolites, making them an important natural resource for producing unique chemical compounds to combat a variety of diseases [
3,
4,
5]. Notably, a multitude of fungal natural products exhibiting various biological effects have been discovered, suggesting that fungi play a role in communicating with other organisms and adapting to different environments [
6,
7]. Some of these identified fungal natural products have already been utilized in the health–functional food, agrochemical, cosmetic, and pharmaceutical industries [
8].
In the early 2000s, technological advances in genome sequencing and bioinformatics on filamentous fungi began to reveal a discrepancy between the number of biosynthetic gene clusters (BGCs) encoding the biosynthesis of fungal secondary metabolites and the actual number of identified fungal compounds from the target strain [
9,
10]. This fact suggested that fungi have a great potential for identifying structurally and/or biologically novel secondary metabolites. However, many BGCs are not actively expressed in the normal laboratory growth environment. These are so-called cryptic or silent BGCs [
11,
12]. It is estimated that there are over 5 million fungal species on earth, and each of these species is capable of producing a variety of secondary metabolites, including bioactive compounds, pigments, and toxins [
9,
12]. These secondary metabolites are produced by specialized biosynthetic pathways, which are encoded by clusters of genes known as BGCs. Despite the availability of over 1000 fully sequenced fungal genomes and the identification of tens of thousands of BGCs, only a small fraction (<3%) of these clusters have been linked to specific secondary metabolites in part because of the cryptic BGCs of fungi [
9].
Neurospora crassa, a member of the Ascomycota phylum, serves as a model organism for the study of fungal genetics, physiology, and development. It has been widely employed to investigate fundamental processes, such as circadian rhythm and gene regulation [
13].
N. crassa is known to produce a variety of secondary metabolites, including carotenoids, melanins, and mycotoxin sterigmatocystin [
13,
14]. The sequencing information of
N. crassa was completed in 2003 and has been found to contain numerous BGCs, many of which are predicted to encode secondary metabolites [
15]. Recently, about 70 BGCs including polyketide synthases (PKSs), non-ribosomal peptide synthetases (NRPSs), terpene synthases, and siderophore synthetases were reported from the sequencing data of the fungus [
14,
15]. However, only a few of BGCs of
N. crassa have been linked to specific secondary metabolites or characterized in detail. Bioinformatics-based predictions of the chemical structures based on the uncharacterized BGCs suggested that many of them were likely to have novel structures. Experimental characterization of these novel metabolites is often challenging since many BGCs are weakly expressed under laboratory conditions and may require specific environmental cues, growth conditions, and extraction and isolation techniques to induce production [
16,
17,
18].
After the completion of genome sequencing on
N. crassa, the genomes of many fungi, including those of both Ascomycota and Basidiomycota phyla, have been found to contain numerous cryptic BGCs [
19].
Aspergillus nidulans is one of the most well-studied secondary metabolite producers.
A. nidulans has been shown to produce a diverse array of secondary metabolites, including emericellamides, terrain, asperfuranone, fumitremorgins, gliotoxin, and aspernidine A [
20]. Several studies have used computational methods to predict the number of BGCs in the
A. nidulans genome. One such study, published in 2015, identified 52 BGCs in
A. nidulans using a combination of genome mining and phylogenetic analysis [
21]. Another study, published in 2018, identified 63 BGCs in the strain using a similar approach [
21].
The discovery of cryptic BGCs in microorganisms, including fungi, has spurred the development of new experimental methodologies for identifying the secondary metabolites of these clusters, which led to the realization that they have the potential to produce novel specialized metabolites, giving rise to a new field of research called genome-guided natural product discovery [
22]. Aside from pinpointing the genomics-driven approach, traditional approaches for identifying and characterizing natural products, such as fractionation and purification followed by structural elucidation using techniques such as NMR spectroscopy and mass spectrometry, can be time-consuming and require large amounts of material. To address these challenges, newer approaches such as metabolomics, transcriptomics, and proteomics have been developed to more efficiently identify and characterize natural products from cryptic BGCs [
23].
2. Organization of Biosynthetic Gene Clusters of Fungi and Their Regulation
Fungi can produce various secondary metabolites with diverse biological activities, such as antibiotics, antifungals, immunosuppressants, and anticancer agents. These secondary metabolites are often encoded by BGCs, which are physically co-localized on the fungal genome and contain all the genes necessary for the biosynthesis of the corresponding secondary metabolite [
21,
24]. The organization of BGCs in fungi can differ depending on the type of secondary metabolite being produced, but there are some common features. Typically, BGCs are composed of a core set of genes that encode enzymes responsible for the biosynthesis of secondary metabolites, as well as regulatory genes that control gene expression and coordinate the biosynthesis process [
24,
25]. In many cases, BGCs are found within mobile genetic elements such as transposable elements, plasmids, or integrative and conjugative elements, which can facilitate their transfer between different fungal strains or even different fungal species. The structure of BGCs can also be highly variable, with some BGCs containing only a few genes, while others can harbor dozens of genes that are organized into sub-clusters or modules. These sub-clusters may be responsible for the synthesis of different parts of the secondary metabolite, which are then combined to form the final product [
26,
27].
Fungal BGCs can be quite large, often exceeding 100 kb in size [
19,
28]. This fact presents a challenge for researchers who want to study the activity of these gene clusters by expressing them heterologously in a different host organism, such as
E. coli or yeast. Fungal BGCs are classified based on the type of secondary metabolite they encode, including polyketides, non-ribosomal peptides, terpenoids, saccharides, and ribosomally synthesized and post-translationally modified peptides (RiPPs) [
29,
30,
31]. The organization of BGCs in fungi is highly complex and dynamic, reflecting the diverse functions and ecological roles of the secondary metabolites they produce. Polyketide synthases (PKSs) are a class of enzymes found in fungi and other organisms that are responsible for the biosynthesis of polyketides. PKSs are modular enzymes that utilize a repeating cycle of catalytic domains to assemble complex polyketide chains from simple building blocks, such as acetate and malonate. Each module typically contains several different domains that are responsible for different steps in the biosynthesis process, such as chain initiation, chain elongation, and chain termination. Fungal NRPs utilize a repeating cycle of catalytic domains to assemble complex peptides from simple amino acid building blocks. Each module in NRPs harbors several various domains, which lead to the biosynthesis processes including amino acid activation, amino acid incorporation, and peptide bond formation.
The position of fungal BGCs is usually observed proximal to the telomeres in the genome and often within heterochromatin regions [
32]. Heterochromatic regions are generally considered to be silent regions of the genome with low gene density and reduced recombination [
32,
33]. This may provide a more stable genomic environment for the BGCs, which are often under positive selection due to their role in fungal survival. The expression of BGCs is tightly regulated via a complex interaction of genetic, epigenetic, and environmental factors. The regulation of BGCs is important for ensuring that these clusters are expressed under appropriate conditions and that the products of biosynthesis are synthesized and utilized efficiently. A transcription factor is a protein that can bind to specific DNA sequences and activate or repress gene expression. Many BGCs are controlled by transcription factors that are specific to the biosynthetic pathway and that respond to environmental signals to activate or repress expression [
19]. The structure of chromatin has a significant impact on gene expression in fungi. The presence of histone modifications such as methylation, acetylation, and phosphorylation controls the accessibility of DNA, and therefore the expression of the genes within BGCs [
34]. Many BGCs are expressed in response to specific environmental triggers, such as nutrient availability or the presence of competing organisms. These signals affect transcription factors or other regulatory elements that modulate the expression of BGCs. In some cases, BGCs can be acquired through horizontal gene transfer, which involves the transfer of genetic material from one organism to another [
35].
Fungi are highly adaptable organisms that live in diverse and complex natural environments, and their growth and metabolism are influenced by a variety of biotic and abiotic factors. However, laboratory growth conditions are usually simple and standardized and may not accurately reflect the physical structure, nutrient availability, and microbial diversity of actual natural environments. Additionally, laboratory conditions may not accurately mimic the environmental stresses that fungi encounter in the wild, such as changes in temperature, pH, osmotic pressure, and competition with other microorganisms. As a result, fungal fermentations in the general laboratory may not accurately represent the full range of metabolic and biosynthetic capabilities that fungi exhibit in their natural habitats [
36]. Therefore, understanding the regulatory processes that modulate the growth, metabolism, and biosynthetic capabilities of fungi is critical for unlocking their full potential as sources of bioactive compounds. This requires a multidisciplinary approach that combines microbiology, biochemistry, and genetics to fully understand the complex regulation of fungi.
3. Characterization of Biosynthetic Gene Clusters and Natural Product Discovery
Next-generation sequencing (NGS) technologies have revolutionized the field of genomics by allowing rapid and cost-effective acquisition of genomic data. The rapid pace of technological advancements in NGS has led to an exponential increase in the amount of fungal genomic data generated, which has in turn fueled the development of new analytical tools and computational approaches to handle and analyze these data [
27,
37]. It is now common for researchers to identify BGCs responsible for the production of fungal secondary metabolites. By obtaining a draft genome sequence of fungi, researchers utilize a variety of bioinformatic tools to identify and analyze potential BGCs involved in the biosynthesis of a particular compound of interest (
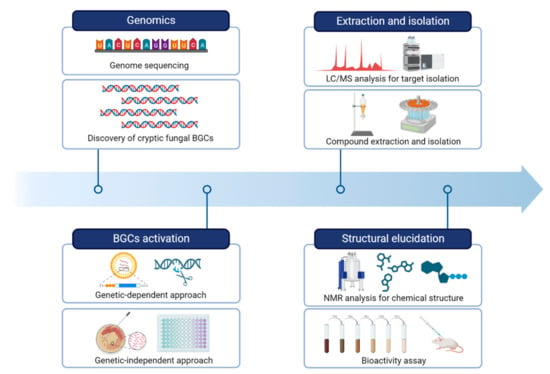
Figure 2).
Figure 2. A workflow of strategies for natural product research by activating fungal BGCs.
Once a draft genome sequence has been acquired, researchers start to use tools such as antiSMASH and MIBiG to identify potential BGCs within the genome [
38,
39]. These tools analyze the genome sequence for specific gene clusters known to be involved in the biosynthesis of secondary metabolites, such as polyketides, non-ribosomal peptides, and terpenes. By comparing the identified BGCs to known BGCs in databases, researchers can predict the structure and function of the secondary metabolites. antiSMASH is one of the most widely used BGC detection tools [
38]. It is a web-based tool that allows users to input draft genome sequences and predict the location of BGCs in the genome. The tool uses a variety of algorithms to identify BGCs, including hidden Markov models (HMMs), Pfam domains, and Clusters of Orthologous Groups (COGs). The tool also provides annotations of the predicted BGCs, including predictions of the chemical structure of the metabolite produced. Alternative tools for BGCs identification are PRISM and BAGEL [
40,
41]. PRISM uses a machine learning algorithm to predict BGCs in microbial genomes. It integrates multiple data sources, including gene co-occurrence patterns, gene expression data, and functional annotations, to identify BGCs. PRISM also includes tools for visualizing and exploring predicted BGCs, including interactive network visualizations. BAGEL is a tool specifically designed for the identification of bacteriocin gene clusters, which are BGCs involved in the biosynthesis of antimicrobial peptides produced by bacteria. BAGEL uses a combination of HMMs and machine learning algorithms to predict bacteriocin gene clusters in microbial genomes.
When the above tools are applied to the genome sequence of a specific fungal strain, they are expected to identify cryptic BGCs that might be responsible for the production of unknown compounds and would be a promising starting point for natural product discovery. The type of NGS technology utilized impacts the capability to characterize the full complement of BGCs in a particular genome [
42]. Different NGS technologies have different strengths and limitations, and the choice of technology will depend on factors such as the size and complexity of the genome, the sequencing depth needed, and the availability of bioinformatics tools and resources [
43]. Some NGS technologies, such as PacBio and Oxford Nanopore, can generate long reads that can span entire BGCs and provide more complete sequence information than short-read technologies such as Illumina [
44]. In addition to the choice of NGS technology, the ability to identify the full complement of BGCs in a particular genome also depends on the quality of the genome assembly, the bioinformatics tools used for BGC identification, and the expertise of the researchers involved [
43,
44]. Genome assembly is a critical step in NGS-based approaches for BGC identification, as errors or gaps in the assembly can lead to missed or incomplete BGCs.
Although NGS-based approaches have been utilized to identify the full complement of putative specialized metabolite BGCs, there are limitations to the application of natural product discovery. It is still challenging to predict the chemical structures of the metabolites produced by these BGCs based only on genomic and bioinformatic information [
45,
46]. The identification of BGCs is just the first step in the process of discovering and characterizing natural products. After a specific BGC is identified, it is necessary to express and characterize the genes involved in the biosynthesis of the secondary metabolite, to produce and purify the metabolite itself, and to test its relevant biological activity. These steps require significant resources and expertise, and may not be feasible for all BGCs identified from genomic data (
Figure 2).
This entry is adapted from the peer-reviewed paper 10.3390/separations10060333