Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
|
Biochemistry & Molecular Biology
Over 1.2 million deaths are attributed to multi-drug-resistant (MDR) bacteria each year. Persistence of MDR bacteria is primarily due to the molecular mechanisms that permit fast replication and rapid evolution. As many pathogens continue to build resistance genes, current antibiotic treatments are being rendered useless and the pool of reliable treatments for many MDR-associated diseases is thus shrinking at an alarming rate. In the development of novel antibiotics, DNA replication initiation and the primosome are still largely underexplored targets.
- DNA replication
- antibiotics
- primase
- helicase
1. DNA Replication: A Valid Antibacterial Drug Target
In Escherichia coli, two large multimeric protein complexes, referred to as replisomes, assemble at a single, unique replication origin (oriC) of the circular chromosome. Each replisome proceeds in opposite direction at approximately 60 kb/min, duplicating the leading and lagging strand until a terminus site is reached (Figure 1A) [1,2,3,4]. In favourable conditions, bacterial DNA replication can occur in an overlapping manner termed multi-fork replication, whereby a second round of replication can begin prior to the first-round finishing (Figure 1A) [5,6]. Multi-fork replication is required for rapid growth of E. coli with generation times as low as ~20 min and the inheritance of partially replicated chromosomes in daughter cells. Regardless, DNA replication must be completed faithfully to guarantee the integrity of the genome. Whilst bacterial chromosome replication is often described based on the well-characterised E. coli system, significant variations do exist in other bacteria [1,3,4,7,8,9,10,11].
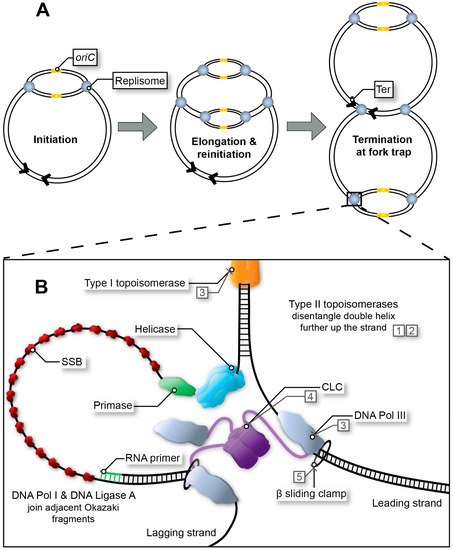
Figure 1. Replication fork progression from initiation to termination. (A) Multi-fork replication in fast growing bacteria. Multiple replisomes may load sequentially at the origin site (initiation at oriC in E. coli), as a result of reduced generation time. (B) Bacterial replisome during fork progression (elongation stage). After the initiator protein (not depicted) has separated the DNA to form the open complex, the helicase unwinds the replication bubble. RNA primers are then synthesised by the primase and extended by DNA Pol III*. Polymerase activity is mediated by the β sliding clamp and its clamp loading complex (CLC). Exposed single-stranded DNA (ssDNA depicted here using a single line) is protected by single-stranded binding protein (SSB) throughout this process. In E. coli, adjacent Okazaki fragments on the lagging strand are joined by DNA Pol I and DNA Ligase A (not depicted). Topoisomerases release tension in the double helix as the fork progresses. Type I topoisomerases achieve this by nicking one strand within the double helix, whereas type II topoisomerases disentangle double-stranded DNA (dsDNA) by cutting both strands and passing one duplex DNA through the other to remove loops created by supercoiling. Examples of inhibitors: quinolones (1), aminocoumarins (2), and experimental compounds with corresponding DrugBank database accession numbers DB01643 (3), DB02930 (4) and DB06998 (5).
2. Targeting DNA Replication Initiation
The initiator protein (DnaA), helicase (DnaB), and primase (DnaG) are recruited to the origin of replication (oriC in E. coli) to create the initial RNA primers that will be extended (leading strand) by the DNA polymerase after replisome assembly (Figure 1B). All three are essential for maintaining the stability of the genome and for the formation of new, viable bacterial cells, and thus have potential to be exploited as targets in drug discovery campaigns [13,15,30]. The helicase and primase are the most important proteins in the primosome that synthesises RNA primers at regular intervals on the lagging strand and for DNA replication restart [31]. While single-stranded DNA binding protein (SSB) and DnaB are sufficient to recruit DnaG [32] to a functional replication fork, other proteins are sometimes necessary when a replication fork has stalled and is ‘abandoned’ [33]. In E. coli this requires a set of DNA replication restart proteins, PriA, PriB, PriC, DnaT along with SSB to reassemble the helicase and primase [31]. This particular system is commonly referred to as the replication restart primosome. Of note, only PriA is conserved across all bacteria. In Mycobacteria, no homolog for any other replication restart protein has been identified [9]. While PriA is essential and kaempferol has been identified as a putative inhibitor of the Staphylococcus aureus protein [34], no further compound showing activity on PriA or other replication restart proteins has been reported.
The bacterial replication initiator, helicase and primase have functional analogs in eukaryotes but share very little sequence homology [35,36]. Thus, they can be safe and specific targets of inhibitors within a mammalian host. Comparatively, the protein sequences of the initiator, helicase and primase are relatively well-conserved amongst bacteria. Of note, the primase shows the greatest divergence in sequence conservation between Gram-negative and Gram-positive bacteria, suggesting its potential as a narrow-spectrum target [37]. The same degree of sequence conservation is not usually observed for other initiation proteins. For example, helicase loading onto single-stranded DNA (ssDNA) can be mediated by different proteins [38], undermining the potential of the helicase-loader proteins as drug targets.
The druggability of the initiator, helicase and primase is evidenced by their capacity to bind and be inhibited by small, drug-like molecules. For example, both DNA and nucleotide triphosphate (NTP) interactions by the Mycobacterium tuberculosis primase were inhibited by competitive binding of suramin and doxorubicin [39]. Flavonols have been shown to inhibit the Klebsiella pneumoniae helicase by competitively binding to its ATP-binding pocket, a similar mechanism to which has been suggested to inhibit the initiator of E. coli by bisindoles [40,41]. NMR screening assays have identified a novel ligand-binding site on the S. aureus primase separate from those previously characterised as cofactor-binding sites, showing potential for non-competitive inhibition of the protein [42]. Overall, these proof-of-principle data establish the initiator, helicase and primase as specific and druggable targets [43].
3. Replication Initiation Inhibitors
The global burden of MDR bacteria is continually increasing [18]. DNA replication offers a multitude of safe and specific drug targets, yet very few have progressed beyond validation studies (Figure 1B). The initiator protein, helicase and primase have been proposed to be attractive targets for drug development; however, development of inhibitors to even a pre-clinical stage is yet to be seen. To date, only preliminary screening studies have been performed to uncover feasible inhibitors targeting the initiation and primosome proteins (Table 1). The details of these inhibitors have been summarised below.
Table 1. Inhibitors targeting the initiator, helicase and primase proteins.
| Protein Target | Inhibitor | Species * | Ref. |
|---|---|---|---|
| DnaA | Bisindole derivatives | Eco | [41,112] |
| DnaB | Myricetin | Eco | [113] |
| Galangin | Kpn | [114] | |
| Coumarin-based | Ban Sau |
[115] | |
| Triaminotriazine derivatives | Eco Sau Pau |
[116] | |
| DnaG | Para-phenyl substituted tetrazoles | Eco | [117] |
| Benzo[d]imidazo[2,1-b]imidazoles Benzo[d]pyrimido[5,4-b]furans Pyrido[3’,2’:4,5]thieno[3,2-d]pyrimidines |
Eco | [118] | |
| Tilorone Doxorubicin Suramin |
Ban | [119] | |
| Doxorubicin Suramin Ellagic acid |
Mtb | [39] | |
| Anthracyclines and aloe-emodin | Mtb | [120] | |
| Daunorubicin derivatives | Mtb | [121] | |
| 3-[1-benzyl-5-chloro-2-(ethoxycarbonyl)-4-(trifluoromethyl)-1H-indol-3-yl]propanoic acid 3-[1-benzyl-7-chloro-2-(ethoxycarbonyl)-5-(trifluoromethyl)-1H-indol-3-yl]propanoic acid |
Mtb | [122,123] | |
| 9-fluorenone-based derivatives | Sau Ban Bth |
[124] | |
| Dequalinium analogues | Sau | [125] | |
| Phenolic monosaccharides derived from Polygonum cuspidatum | Eco | [126] | |
| Bicyclic 10-membered macrolide (Sch 642305) from Penicillium verrucosum |
Eco | [127,128,129] |
* Eco—Escherichia coli; Kpn—Klebsiella pneumoniae; Ban—Bacillus anthracis; Sau—Staphylococcus aureus; Pau—Pseudomonas aeruginosa; Mtb—Mycobacterium tuberculosis; Bth—Burkholderia thailendensis.
4. Advantages and Limitations of Current Methods
The mechanisms of DNA replication initiation and associated protein–protein interactions (PPI) have potential as targets for the development of safe and effective antibiotics (Table 1). Though this has been repeatedly recognised over the past 10 years [9,14], limited progress has been made towards clinical trials for hits targeting bacterial initiator, helicase, and primase proteins [30]. Largely, this is due to the formal characterisation of these initiation and primosome proteins historically being limited to the model bacteria, E. coli and Bacillus subtilis, and some ESKAPE pathogens (Enterococcus faecium, S. aureus, K. pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterococcus spp). Indeed, several essential DNA replication proteins and PPIs have been found to differ significantly between the E. coli model and other bacteria, limiting broad-spectrum application [3,132]. In addition, there are other compounding issues associated with screening assays targeting replication processes involving multiprotein complexes, e.g., difficulty to decipher the specific mechanism of action of a hit, and failure to demonstrate in vivo activity due to various target access barriers (Table 2).
While most in silico and in vitro screening methods fail to identify compounds that demonstrate in vivo activity, cell-based assays cannot identify a direct mechanism of action or ensure selectivity. Table 2 presents a selection of promising methods available to characterise the functions and interactions of the proteins that have been or can be used to screen libraries of compounds.
Table 2. Screening assays for the initiator, helicase and primase proteins.
| Target | Species * | Type | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|---|
| DnaA | Eco | Cell-based (dnaA219rnhA reporter strain) | Compounds can cross bacterial membrane | Cannot distinguish compounds with specificity for DnaA | [133] |
| Eco | Minichromosome-based (GFP reporter) | Compounds can cross bacterial membrane | Inhibition of plasmid-based oriC not always repeatable with chromosomal oriC | [134] | |
| Eco | Cell-based (pBR322-DARS2 and hda mutant reporter strains) | Compounds can cross bacterial membrane | Cannot distinguish compounds with specificity for DnaA | [135] | |
| Eco | Filter binding assay ([α-32P]ATP) | Could be converted for HTS | Requires radioactive labelling and scintillation counter | [41] | |
| Spy | In silico (molecular dynamics simulation) | Inexpensive, can be used for pre-screening | Requires additional in vivo efficacy conformation | [136] | |
| Eco | Cell-based (SF53 reporter strain) | HTS format (384-well plates) | Cannot distinguish compounds with specificity for DnaA | [137] | |
| DnaB | Kpn | ATPase assay (molybdophosphoric acid complex) | Could be converted for HTS | Low throughput | [40] |
| Kpn | Helicase activity assay (FRET 1) | Could be converted for HTS | Low throughput | [40] | |
| Sau | ATPase assay (molybdophosphoric acid complex) | HTS format (96-well plates) | Requires helicase activity assay for confirmation | [115] | |
| Eco Sau Ban Pau |
Helicase activity assay (FRET 1) | HTS format (96-well plates) | Cannot distinguish compounds with specificity for DnaB | [115,116,138,139,140] | |
| Kpn | dNTP dissociation (fluorescence) | Could be converted for HTS | Requires helicase activity assay for confirmation | [114] | |
| viral and bacterial | Time-resolved FRET 1 (Tb3+, Eu3+) | HTS format (Up to 1536-well plates) | Optical interference from compounds | [141] | |
| Eco Bst |
ATPase assay (NADH) | Could be converted for higher throughput | Low throughput | [113,132] | |
| Bst | Helicase activity (radioactive label not specified) | Can be used for inhibitors of DnaB/G interaction | Low throughput, requires radioactive labelling | [132] | |
| UvrD | Eco | Helicase activity (SYTOX stain) | Microfluidic flowcell format, could be used for DnaB | Low throughput | [142] |
| DnaB/ DnaG |
Bst | Reverse yeast three-hybrid (β-galactosidase) | Allows screening of potential antimicrobial peptides | Low throughput, for screening of peptides only | [143] |
| Eco | SPA 2 ([3H]CTP) | HTS format (96-well plates) | Requires radioactive labelling and scintillation counter | [118,138] | |
| DnaG | Eco | Thermally denaturing HPLC (260 nm) | Can distinguish between de novo synthesis and elongation | Low throughput | [113,144] |
| Mtb Ban Sau |
Pyrophosphatase assay (molybdophosphoric acid complex) | HTS format (384-well plates) | Cannot distinguish compounds with specificity for DnaG | [39,119,120,125] | |
| Eco Mtb |
Primase activity ([3H]NTP, [α-32P]ATP) | Can be used for inhibitors of DnaB/G interaction | Low throughput, requires radioactive labelling, cannot distinguish compounds with specificity for DnaG | [123,126,128] | |
| Eco | SPR 3 competition assay | Can be used for inhibitors of DnaG/SSB interaction | Cannot distinguish compounds with specificity for DnaG | [117] | |
| Eco T7 |
STD 4 and 2D NMR (Imax, 15N-1H HSQC 5) | Could be used for inhibitors of other proteins | Requires pooling of compounds for initial screens | [117,122] | |
| Eco | Primase/Replicase activity assay (PicoGreen) | Can be used for inhibitors of DnaG/SSB interaction, HTS format (384-well plates) | Cannot distinguish compounds with specificity for DnaG | [145,146] | |
| All | Any species | Molecular docking (AutoDock Vina, Glide, Molecular Operating Environment) |
Can give indication of mechanism of action | Requires additional in vivo efficacy conformation | [117,118,121,122,136] |
| Any species | SPR 3 | Sensitive, can determine binding kinetics | Expensive, need to control for buffer effects | [147,148] | |
| Any species | Mass spectrometry (AS-MS 6, LC-ESI-MS 7) |
Sensitive, can pool compounds to increase throughput | Requires multiple rounds for inhibitor ranking | [147,149] | |
| Any species | Thermofluor (SYPRO Orange & ANS stain) | HTS format (384-well plates) |
Optical interference from compounds | [147,150] | |
| Any species | DSF-GTP 8 (GFP) | HTS format (96-well plates), can be used in mixed samples, can test target access |
Optical interference from compounds | [151,152] |
* Eco—Escherichia coli; Spy—Streptococcus pyogenes; Kpn—Klebsiella pneumoniae; Sau—Staphylococcus aureus; Ban—Bacillus anthracis; Pau—Pseudomonas aeruginosa; Bst—Bacillus stearothermophilus; Mtb—Mycobacterium tuberculosis; T7—T7 bacteriophage. HTS: High-throughput screening. 1 Fluorescence resonance energy transfer; 2 Scintillation proximity assay; 3 Surface plasmon resonance; 4 Saturation transfer difference; 5 Heteronuclear single quantum coherence; 6 Affinity selection-mass spectrometry; 7 Liquid chromatography-electrospray ionisation-mass spectrometry; 8 Differential scanning fluorimetry of GFP-tagged proteins.
Many activity assays used in past drug screening initiatives for replisome proteins were performed in low throughput, such as the [α-32P]ATP-based filter binding assay for E. coli DnaA (and primosome) developed by Mizushima et al. [41] and the thermally denaturing HPLC primer synthesis assay for E. coli DnaG developed by Griep et al. [113]. In some cases, low-throughput activity assays could be adapted to a high-throughput format, as seen with the helicase activity assays used by Lin and Huang [40] where both the ATPase and 5′-3′ DNA helicase activity assays have been adapted to 96-well plate formats for helicases from E. coli [138], Bacillus anthracis and S. aureus [115,140]. The filter binding assay developed for the E. coli primosome [41] is another example that could be adapted to a 96-well plate format using a cell harvester and filter plates. More recent activity assays have been developed with higher throughput formats, including the coupled colorimetric primase–pyrophosphatase assay developed by Biswas et al. for M. tuberculosis DnaG [39,119] and the GFP reporter-based minichromosome assay developed by Klitgaard et al. for E. coli DnaA [134].
Most DNA replication initiation inhibitors have yet to demonstrate activity in in vivo trials [40,119,140,141,143]. For example, inhibitors of Bacillus stearothermophilus helicase–primase interaction [143] identified with a reverse yeast three-hybrid assay were unable to demonstrate antibacterial activity [30]. In another example, in vitro inhibitors of S. aureus and B. anthracis helicase identified with a fluorescence resonance energy transfer (FRET)-based assay demonstrated low activity in vivo, with insufficient inhibition to obtain MIC values [140]. Furthermore, specific inhibitors of B. anthracis primase identified with the coupled colorimetric primase–pyrophosphatase assay also failed to show in vivo activity, due to an inability to penetrate the bacterial envelope [119]. More recent studies applying molecular docking approaches [121,136] and/or fragment-based screens [117,122] have identified inhibitors with their activity yet to demonstrate in vivo. Multi-target approaches increase the potential for identification of lead compounds [128]. Dallmann et al. [145] used high-throughput parallel multiplicative target screening approach building on a PicoGreen fluorescence-based assay for the screening of E. coli and B. subtilis replisome. However, as these include multiple proteins, identifying the mechanism of action can be challenging.
One other major issue with these assays is the possibility of non-specific DNA interaction leading to assay failure. For example, Biswas et al. [119] identified doxorubicin as an inhibitor of B. anthracis primase with bacteriostatic effects in vivo, yet were unable to determine its direct mechanism of action. Doxorubicin is a DNA intercalator which could explain its activity [153]. Another example of this can be seen in the use of the assay developed by Fossum et al. [133] for identifying inhibitors of DnaA in E. coli, instead detecting inhibitors of DNA gyrase [137]. Furthermore, deferoxamine identified using a cell-based assay with an E. coli ATP-DnaA-‘locked’ strain [135] failed to restrict the growth of wild type cells and was found to act via iron-chelating activity. Unfortunately, these potential complications arise in many current biochemical activity assays for replisomal proteins when DNA is essential to their operation, leading to the inability to distinguish whether inhibition is due to targeting of the enzyme or interactions with the DNA itself.
In the last two decades, large scale drug screening campaigns have shifted to using methods that can detect the physical interaction of a compound with its protein target [147,149,154]. Fragment-based approaches involving high-throughput biophysical screening techniques have become a common feature in drug discovery. High-throughput surface plasmon resonance (SPR) is now sensitive enough to be used as a primary screen [155]. Of note, the interactions of helicases and primases have been characterised by SPR validating the utility of this technique for compound screening with these proteins [78,101]. In addition, SPR can be used for subsequent validation and kinetic characterisation of the interactions. However, stability of the target can be problematic and the cost of screening is high; both of which are important aspects to consider [147,148,155]. Specialised MS techniques to screen small molecules [156,157,158] include affinity selection-mass spectrometry (AS-MS) and pulsed ultrafiltration-mass spectrometry (PUF-MS) [159,160]. They are suitable for high-throughput screening of large compound libraries or natural product extracts on protein targets [161,162]. The helicase has been examined by native MS [163,164] and as such, should be amenable to AS-MS. Differential scanning fluorimetry (DSF) [150,165], also known as Thermofluor, is probably the most-commonly used technique as a first step in the process of large compound library screening due to its technical simplicity and low cost. A derivative of this technique, DSF of GFP-tagged proteins (DSF-GTP), has been validated with several E. coli and Burkholderia pseudomallei GFP-tagged replisomal proteins such as Tus, DnaA, DnaB and DnaG [151,152,166,167]. DSF-GTP has several advantages over classic DSF in that it can be used with protein mixtures and extracts to evaluate target access [152], providing a powerful platform for future large scale drug screening campaigns.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24108802
This entry is offline, you can click here to edit this entry!