Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Paraneoplastic neurological syndromes (PNS) include any symptomatic and non-metastatic neurological manifestations associated with a neoplasm. PNS associated with antibodies against intracellular antigens, known as “high-risk” antibodies, show frequent association with underlying cancer. PNS associated with antibodies against neural surface antigens, known as “intermediate- or low-risk” antibodies, are less frequently associated with cancer.
- paraneoplastic neurological syndromes
- central nervous system
- neurology
- oncology
- movement disorders
- ataxia
- epilepsy
- immune-checkpoint inhibitors
1. Introduction
Paraneoplastic neurological syndromes (PNS) of the central nervous system (CNS) consist of neurological manifestations associated with a neoplasm unrelated to the direct invasion/metastasis in the CNS [1][2]. PNS complicates approximately 1–15% of cancers, varying with the associated cancer type [1][3][4]. PNS may precede the diagnosis of cancer by 1 to 5 years in up to 70% of patients [5][6]. PNS are thought to arise from an immune response directed against common antigens/epitopes shared by tumor cells and normal healthy cells within the CNS [7]. Conversely, in non-neurological paraneoplastic syndromes and in PNS of the peripheral nervous system, the target antigen is located outside the CNS. When PNS involve the peripheral nervous system, they cause myopathies and/or myasthenia-gravis-like syndromes. Details regarding the PNS of the peripheral nervous system have been further explored in another article in this issue. PNS of the CNS, despite being relatively rare, have been increasingly recognized and detected over time, with a constantly growing body of newly discovered antibodies and expanded interest among clinicians over the last few years [8]. Hence, PNS of the CNS are often considered in the differential diagnoses in patients presenting with acute or subacute encephalopathy once the more common causes (e.g., infections, toxic and metabolic conditions, and even functional neurologic disorders or psychiatric disorders) are excluded [9][10].
At the cellular level, PNS-associated cancers may harbor gene variants coding for onconeural proteins, particularly highly immunogenic antigens that are also expressed by CNS cells and which activate the immune system [11]. Antibodies directed against intracellular (e.g., cytoplasmic, nuclear, or synaptic) neuronal antigens are traditionally named “onconeural” antibodies and are associated with cytotoxic T cells (which are thought to exert a pathogenic role) [12]. Indeed, despite their relevant role as biomarkers, these antibodies do not have a direct pathogenic role. Conversely, antibodies against neuronal surface antigens (NSA-Abs) have a direct pathogenic role but they are less likely to be associated with cancer (they are the expression of immune system activation within the context of a systemic immune condition or disease). NSA-Abs are directed against ion channels, receptors, or other components of neural membranes [1]. The distinction between onconeural and NSA-Abs has therapeutic implications: immune treatments can be highly effective for PNS associated with NSA-Abs, while they are less effective in PNS associated with onconeural antibodies (See Figure 1).
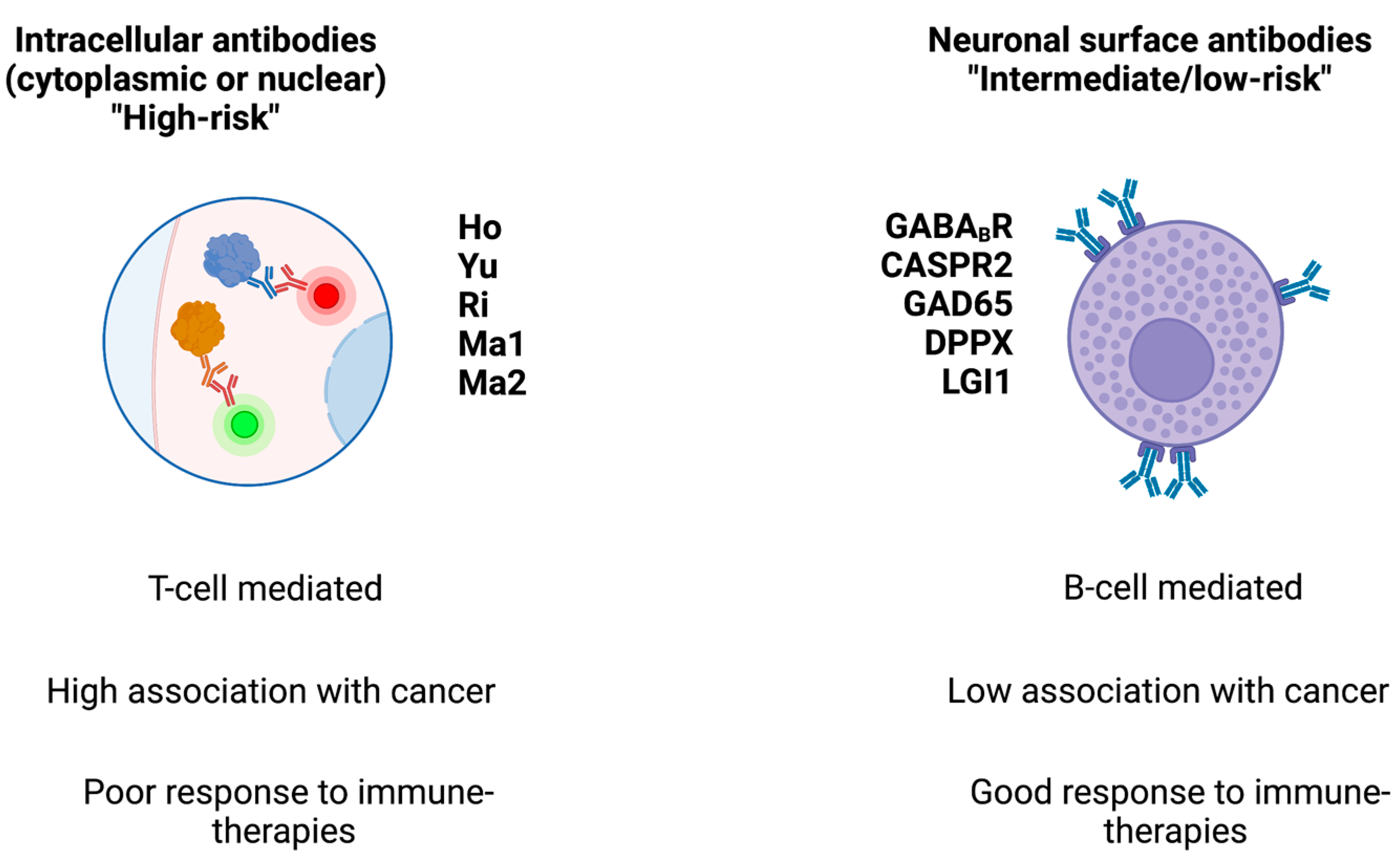
Figure 1. Main classes of antibodies, their associated cancer risk, and response to therapies. The figure schematizes the two main classes of antibodies found in paraneoplastic syndromes.
2. Rapidly Progressive Cerebellar Syndrome
Rapidly progressive cerebellar syndrome, previously known as subacute cerebellar degeneration, results from inflammation-mediated degeneration of cerebellar Purkinje cells leading to ataxia becoming severely disabling under three months [1][13][14]. Hyperacute and delayed presentations have also been described [15]. Ataxia typically manifests with gait abnormalities followed by truncal and appendicular ataxia [16]. Additional brainstem involvement includes dysarthria and oculomotor abnormalities [16][17]. Imaging is often unremarkable early in disease course as radiographic evidence of cerebellar atrophy appears in late stages [1].
While rapidly progressive cerebellar syndrome has been linked to “high-risk” antibodies, “intermediate- and low-risk” antibodies are being increasingly implicated. One of the best described antibodies is Anti-Yo (also known as Purkinje cell antibody (PCA)—1). Anti Yo/PCA1 is considered a “high-risk” antibody because it is highly associated (>90%) with cancer, typically ovarian or breast. It presents with a cerebellar syndrome, often preceded by a prodromal period of vertigo [18][19][20]. Tr/delta/notch-like epidermal growth factor-related receptor (DNER) is another “high-risk” antibody with >90% association with cancer, typically Hodgkin lymphoma [16][21][22]. Anti-Ri/ANNA-2 is a ‘high-risk’ antibody (>80% association with cancer, primarily breast) that is classically linked to OMS (See also Section 3), but the current literature suggests it is likely more commonly associated with rapidly progressive cerebellar syndrome [23]. Less frequently, anti-Ri/ANNA-2 antibodies may present as Bickerstaff brainstem encephalitis and with oculomotor abnormalities suggestive of progressive supranuclear palsy (PSP); however, with sudden onset or stepwise or rapid progression (to differentiate it from the classic form of PSP). [23]. More recently, KLHL11 ab has been identified as an additional “high-risk” antibody (>80% association with testicular seminoma) that causes an overlapping progressive cerebellar and brainstem syndrome, typically accompanied by sensorineural hearing loss [24][25]. This constellation of symptoms is captured with the MATCH criteria where points are allocated for male sex (1 point), ataxia (1 point), testicular cancer (2 points), other type of cancer (1 point), and hearing disturbances (1 point). This validated scoring system has high sensitivity and specificity (78% and 99%, respectively) with scores of ≥4 [26].
Beyond the above, two antibodies which have been classically associated with Lambert–Eaton myasthenic syndrome (LEMS, not a focus of this research), SOX-1 and P/Q voltage gated calcium channel (VGCC), have also been implicated in progressive cerebellar ataxia [27]. LEMS and cerebellar ataxia may co-occur in patients affected by these antibodies and the presence of ataxia portends a much higher likelihood or paraneoplastic origin than LEMS alone [28]. Finally, a few additional antibodies have been reported in paraneoplastic cerebellar syndromes. These include PCA2, mGluR1, antibodies to the intracellular 65-kD glutamic acid decarboxylase (GAD65) enzyme, anti-collapsin-response-mediated protein 5 (CRMP-5), anti-amphiphysin, anti-ANNA-3, dipeptidyl-peptidase-like protein 6 (DPPX), IgLON5, and contactin-associated protein-like 2 (CASPR2) [1][16]. It is quite possible that progressive cerebellar syndromes can be explained by the presence of these antibodies, though their descriptions are not as classic as those listed above and alternative diagnoses or false-positive results should be considered.
Recognition of alternative causes of subacute ataxia is vital as many are reversible if identified early in course. Important diseases on the differential include autoimmune processes related to thyroid disease, diabetes, gluten intolerance (celiac disease), Sjogren’s syndrome, toxic/metabolic syndromes such as vitamin deficiencies (B and E) or metronidazole toxicity, infectious processes such as varicella cerebellitis, and prionopathies [29][30][31][32][33][34]. Depending on presentation, a thorough evaluation of the aforementioned entities should be considered in patients with subacute ataxia.
Recently, Manto and colleagues have proposed the new concept of latent autoimmune cerebellar ataxia (LACA) in analogy with the latent autoimmune diabetes in adults (LADA) to underline the subtle disease course of immune-mediated ataxias, including PCD [35]. LADA is a form of type II diabetes mellitus with autoimmune features, the serum biomarker (anti-GAD antibody) is not always present or can fluctuate and tends to progress with a slow pattern [35]. The disease inevitably progresses until complete pancreatic beta-cell failure within a few years. Due to the unclear autoimmune profile, it is difficult to achieve the diagnosis in the early phase before insulin production is altered [35]. LACA has some analogies with LADA, it has a slow progression, paucity of clear-cut autoimmune features, with significant difficulty for the neurologist to achieve the diagnosis in absence of positive and significantly elevated antibody titers [35]. There are some subtle neurological features which could help clinicians towards the autoimmune and paraneoplastic nature of the cerebellar syndrome, in the early phase, before PCD overtly manifests [35]. These features, namely the cognitive fluctuation within the same day (personal observation of the authors), the presence of dizziness/vertigo, vomiting, nausea, and subtle imbalance sensations may present months before the onset of the overt PCD, and clinicians should be aware of these clinical features to warrant an early diagnosis and treatment [35].
3. Opsoclonus–Myoclonus Syndrome
OMS is a rare syndrome for which a diagnosis of definite PNS can be made without the presence of an antibody [16][36][37][38]. Clinically, OMS is described by opsoclonus (conjugate fast and multidirectional saccades without intersaccadic pauses), non-epileptic myoclonus and, variably, ataxia. With the latter, it is referred to as the triad of “opsoclonus–myoclonus–ataxia” [39]. The proposed diagnostic criteria include at least three of the following four findings: (1) opsoclonus, (2) myoclonus and/or ataxia, (3) behavioral change and/or sleep disturbance, and (4) neoplastic conditions and/or presence of antineuronal antibodies [39]. These criteria allow for flexibility in atypical presentations of OMS which can have delayed onset of opsoclonus or myoclonus and markedly asymmetric ataxia [40].
OMS is more commonly seen in the pediatric population where the syndrome is associated with neuroblastoma [41]. Neuroblastoma is detected in over 50% of pediatric OMS cases [42][43]. Despite this well described association between OMS and neuroblastoma, a specific associated antibody has yet to be elucidated. However, a neuroinflammatory process is suspected, given the cytokine and lymphocyte alterations in the cerebrospinal fluid (CSF) of these patients and the presence of CD20+ B lymphocytes and CD3+ T lymphocytes in the tumor microenvironment of OMS-associated neuroblastomas [44]. Additional suspected causes of pediatric OMS include para-infectious (e.g., varicella, influenza, human herpesvirus 6, SARS-CoV-2) inflammatory syndromes as well as familial/genetic neuro-inflammatory syndromes such as Aicardi–Goutières syndrome [40][45][46][47]. A distinguishing feature of adult-onset OMS, as compared to the pediatric population, is that it is more commonly idiopathic (~61%) than paraneoplastic (~39%) [37]. Unlike the pediatric population, in which the paraneoplastic OMS is associated mostly with neuroblastoma, the adult-onset OMS, when paraneoplastic, is associated with breast cancer, ovarian cancer, and small-cell lung cancer (SCLC). In young women, cases have also been linked to ovarian teratoma [16][39]. Additional OMS-associated antibodies reported in the literature include anti-Hu/antineuronal nuclear antigen type 1 (ANNA-1), anti Yo/PCA1, anti-Ma2, and anti-NMDAR [39]. Similar to the pediatric population, para-infectious etiologies are considered the most likely cause of non-paraneoplastic OMS.
4. Paraneoplastic Encephalitides
Brainstem encephalitis is characterized by prominent brainstem involvement followed or not by multisystem neurologic dysfunction (e.g., in association with more widespread encephalitis or rapidly progressive cerebellar degeneration, as discussed above in Section 1) [16][48]. Paraneoplastic brainstem encephalitis can present with a wide array of oculomotor abnormalities, including but not limited to vertical gaze paresis, internuclear ophthalmoplegia, nystagmus, as well as bulbar weakness and dystonias [49]. In the absence of other classical signs of PNS, it can be confused with other brainstem-localizing neurological syndromes, or with PSP-cerebellar subtype (although, in this latter case, the disease progression is slower, over years) [23][50][51][52][53].
Anti-Ri/antineuronal nuclear autoantibody type 2 (ANNA-2) and anti-Ma2 encephalitis are the “high risk” onconeural antibodies most frequently associated with brainstem encephalitis; anti-KLHL11 can be also found but less frequently [16][54]. Anti-Ri/ANNA-2 commonly presents with ataxia but also with oculomotor dysfunction including OMS and vertical gaze paresis. Abnormal movements include myoclonus in about a third of patients, parkinsonism, and cervical and jaw dystonias [23][50]. Parkinsonism with accompanying supranuclear gaze palsy and cognitive impairment has been described in several cases [23][55][56][57][58]. Taken together, Anti-Ri associated-brainstem encephalitis can mimic neurodegenerative disorders in the spectrum of parkinsonism and/or dementia with prominent brainstem and cerebellar involvement [59].
The clinical presentation of anti-Ma2 encephalitis can be highly variable. In contrast to anti-Ri/ANNA-2 antibodies, which are more common in females and highly associated with breast cancer, Ma2 reactivity is found predominantly in men and frequently linked to testicular cancer [23][56][60][61]. Along with similar oculomotor abnormalities (opsoclonus, gaze palsies), many patients develop concomitant cerebellar ataxia or diencephalic symptoms, such as excessive daytime sleepiness, cataplexy, and endocrine dysfunction [60][62][63]. More recently, cases of anti-Ma2 encephalitis have been described with a motor syndrome characterized by proximal muscle weakness, head drop, and bulbar symptoms. Rarely, patients may develop atrophy or fasciculations in the upper extremities mimicking motor neuron disease. T2/FLAIR hyperintensities may be seen on brain MRI in the corticospinal tract [60]. Concomitant Ma1 antibodies are associated with worse outcome, with more frequent brainstem involvement and ataxia; they are more common in women and in those with non-germ cell tumors [56].
Limbic encephalitis classically presents with the subacute onset of neuropsychiatric symptoms, including memory deficits, mood dysregulation, and behavioral changes, and is frequently associated with seizures [64]. Brain MRIs often show T2/FLAIR hyperintensity in the temporal lobes with corresponding EEG findings of localized epileptiform activity [16][56][65]. Of the “high-risk” antibodies associated with limbic encephalitis, anti-Hu/ANNA-1 is the most notable. Anti-Hu/ANNA-1 antibodies are also robustly linked to SCLC in the vast majority of cases, and the associated encephalomyelitis syndrome can cause both central and peripheral nervous system dysfunction. Of note, encephalomyelitis usually occurs with clinical impairment at various sites of the central and peripheral nervous system, also including the dorsal root ganglia, the peripheral nerve, and/or the nerve roots [16]. It is commonly associated with sensory neuropathy/neuronopathy, dysautonomia, intestinal pseudo-obstruction, as well as brainstem, cerebellar, and limbic/cortical encephalitis [66][67][68]. Documented non-SCLC malignancies have included neuroendocrine tumors, adenocarcinomas, squamous cell carcinomas, germinomas, and large-cell tumors, although these represent a minority [16][69][70]. Interestingly, a small series of eight children with anti-Hu/ANNA-1 antibodies described six cases of limbic encephalitis with negative malignancy workup, and another two cases of opsoclonus–myoclonus with underlying neuroblastoma [71].
Anti-NMDA receptor encephalitis is the most well-described of the autoimmune encephalitides, classically associated with teratoma (usually ovarian). Indeed, Dalmau and colleagues first described serum and CSF antibodies to the NR2B and NR2A subunits of the NMDAR in this population [72]. As observed in the larger cohort studies, the median age of onset is in the third decade of life with a strong female predominance of around eighty percent, unsurprising given the teratoma association [73][74]. Of 577 patients treated internationally who had CSF samples analyzed at the University of Pennsylvania or the University of Barcelona, 38% had an associated neoplasm (including nearly half of all females), and ovarian teratoma comprised 94% of these tumors. Extraovarian teratomas accounted for an additional 2% of the total [73].
Various malignancies have also been associated with anti-NMDAR encephalitis, most frequently in middle-aged or elderly patients, and more rarely in children. There have been reports of lung, breast, uterine, and testicular cancers as well as Merkel cell carcinoma, papillary thyroid carcinoma, renal cell carcinoma, and neuroblastoma [73][74][75][76][77]. Interestingly, Bost and colleagues were able to detect expression of the NR1 subunit of the NMDAR in five of eight tested tumor samples, including two immature teratomas, a pineal germ cell tumor with a mature teratoma component, pancreatic neuroendocrine tumor, and prostatic adenocarcinoma, suggesting an underlying mechanism for CNS autoimmunity [74]. Importantly, herpes simplex virus type 1 (HSV-1) is a known non-paraneoplastic trigger of anti-NMDAR autoimmunity, particularly after HSV encephalitis, and should be considered in the appropriate clinical context [78][79][80][81]. The underlying mechanism of this is beyond the scope of this research.
In adults, anti-NMDAR encephalitis begins with a prodrome of mood changes and positive psychotic features, such as hallucinations and delusions. In the acute phase, more severe psychiatric symptoms and memory changes become evident as well as seizures and movement disorders. Dysautonomia and central hypoventilation can be seen later in the course [1][73]. Seizures are present in about eighty percent of patients, both generalized and focal, with about half of patients developing status epilepticus. Half of these cases may be refractory or super-refractory [82][83]. The EEG in around a quarter of these patients shows extreme delta brush, characterized by diffuse, continuous rhythmic delta activity with superimposed fast activity [83]. Highly specific for anti-NMDAR encephalitis, this finding may be a poor prognostic marker, associated with prolonged hospitalization, increased disability, and higher risk of mortality. However, its prognostic value has not been consistent across all studies [84].
Movement phenomenology is frequently hyperkinetic in earlier stages, including classic orofacial and limb dyskinesias, chorea, opisthotonos. Other symptoms may follow, especially rigidity, slowness, or catatonia [1]. Children are more likely to have movement abnormalities earlier in the clinical course, which may be of unexpected phenomenology in comparison to the adult phenotype (e.g., ataxia) [73][85]. Clinical presentation does not appear to vary significantly between paraneoplastic and non-paraneoplastic anti-NMDAR encephalitis; patients with malignancy exhibit worse survival due to the cancer itself [73]. Patients without an underlying tumor to treat may be more likely to relapse [73].
In general, “intermediate-” and “low-risk” antibodies predominate among limbic encephalitides [16]. One of the most common, after anti-NMDAR, is anti-leucine-rich glioma-inactivated 1 (LGI1) encephalitis, which along with CASPR2 is part of the voltage-gated potassium channel complex (VGKCC). These antibodies may be seen alone or in combination, typically in males in the sixth or seventh decade of life [86][87][88]. Distinctive facio-brachial dystonic seizures, which are very brief in duration and occur up to 100 times per day (usually alternating from one side to the other of the body), can occur in nearly half of those with anti-LGI1 encephalitis, and usually precede cognitive symptoms, of which memory deficits are most common [89][90][91]. Seizures occur in the majority of patients with LGI1 antibodies, and radiologic evidence of mesial temporal sclerosis can be found late in the disease course [89].
Antibodies to CASPR2, and to a lesser extent, LGI1, are associated with peripheral nerve hyperexcitability (manifesting as neuropathic pain and neuromyotonia), as well as Morvan syndrome, which also features prominent neuropsychiatric symptoms, dysautonomia (especially hyperhidrosis and hemodynamic instability), and disordered sleep leading to a state once classically described as “agrypnia excitata” [86][87][92][93][94]. Those with reactivity to CASPR2 frequently present with limbic symptoms at onset, more often seizures than cognitive dysfunction, and most have limbic involvement at some point during their clinical course. Cerebellar ataxia is also common in this population [88]. Onset of symptoms can be chronic and progressive, and the presence of MRI abnormalities can be unreliable, thus patients often do not meet criteria for autoimmune limbic encephalitis [86][88][95][96]. The most common neoplastic association with VGKCC antibodies is thymoma, particularly with CASPR2 antibodies compared to LGI1, and an association with acetylcholine-receptor-antibody-positive myasthenia gravis is well-documented [86][87][97][98][99].
Other “intermediate-risk” antibodies found in limbic encephalitis are gamma-amino butyric acid receptor, type B receptor (GABABR) and α-amino-3-hydroxy-5- methyl-4-isoxazolepropionic acid receptor (AMPAR).
Anti-GABABR encephalitis most frequently presents as a limbic encephalitis characterized by marked seizure activity compared to other encephalitides [100][101][102]. Seizures in anti-GABABR encephalitis are more likely to be tonic–clonic in comparison to other encephalitides and more likely to cause status epilepticus and refractory status epilepticus [102]. Half or more of these cases are found to have SCLC [16][100][101]. These patients tend to be older than those with non-paraneoplastic anti-GABABR encephalitis, and may be more likely to present with a “classic limbic syndrome” rather than with OMS or other atypical symptoms. Anti-AMPAR encephalitis presents similarly, with most developing limbic encephalitis and some with clinical or radiological evidence of more widespread cerebral involvement [103]. Like anti-GABABR encephalitis, the majority of cases are paraneoplastic, and most cases are associated with SCLC. However, anti-AMPAR encephalitis is more prevalent in female patients, and other tumor types can be observed, such as thymoma, breast cancer, and teratoma [101][104]. Of note, both GABABR and AMPAR antibodies have been documented with other SCLC-associated antibodies, such as SOX-1 and amphiphysin [101][105].
5. Stiff-Person Spectrum Disorders
SPSD include stiff-person syndrome (SPS) as well as the single-limb (arm or leg) variant, termed stiff-limb syndrome (SLS), and progressive encephalomyelitis with rigidity and myoclonus (PERM). SPSD are most commonly associated with systemic autoimmunity rather than a specific malignancy. Classic SPS, characterized by progressive, lower-extremity- and truncal-predominant muscle stiffness and stimulus-sensitive muscle spasms, is most frequently associated with the “low-risk”anti-GAD65 antibodies [106][107]. GAD65 antibodies may also be associated with cerebellar ataxia, seizure, and/or limbic encephalitis; these syndromes may co-occur with SPSD [108]. Anti-GAD65 antibodies are closely linked to type 1 diabetes mellitus, autoimmune thyroid disease, celiac disease, and other autoimmune conditions, although rare malignancies have been reported in the literature [106][109]. However, other antibodies associated with SPSD can be paraneoplastic in origin, most notably amphiphysin antibodies. Underlying breast cancer is frequent in this population, which tends to be older and mostly female. Cases typically lack the lower limb predominance of classic SPS and instead have more diffuse involvement, including cervical [110]. Amphiphysin antibodies can also be associated with SCLC; in these cases, other SCLC-associated antibodies such as CRMP5, Hu/ANNA-1, or others may coexist. Interestingly, patients with SCLC may be less likely to develop SPS than those with breast cancer [111].
Patients with antibodies associated with PERM, namely glycine receptor (GlyR) and DPPX antibodies, have a lower risk for an underlying malignancy [16]. PERM varies from other forms of SPSD given the presence of myoclonus and, in many cases, hyperekplexia (exaggerated startle reaction to auditory and tactile stimuli), brainstem dysfunction, and dysautonomia, which may include thermoregulatory abnormalities and diarrhea. The latter is a feature of myoclonus or hyperekplexia associated with DPPX [112][113][114]. In severe cases, PERM may even lead to respiratory failure requiring mechanical ventilation [112][113][114]. While the majority of these cases are idiopathic, multiple cases of newly diagnosed or previously treated thymomas and B-cell lymphomas have been reported with anti-GlyR-associated PERM [112][115]. DPPX antibodies have also been linked to B-cell lymphomas [114].
6. Immune-Checkpoint Inhibitors, CAR T-Cell Therapies, and Related Syndromes
ICIs have completely transformed cancer treatments, thus allowing increased survival and better prognosis of numerous solid malignancies [116]. The rationale of ICIs is to boost the immune system against cancer cells (e.g., blocking the immune-checkpoint receptors PD-1 and PDL-1 on the surface of immune cells or tumor cells, respectively) (Figure 2) at the expense of immune-related adverse events (irAEs). IrAEs are generated by the inhibition of negative regulators of the immune system to primarily enhance the antitumor immunity. Hence, ICIs cause adverse effects which resemble autoimmune conditions affecting several organs and systems, including the CNS. IrAEs may involve the cardiac, integumentary, endocrine, gastrointestinal, hematological, pulmonary, renal, and musculo-skeletal systems [117]. The prevalence of neurological irAEs (n-irAEs) is highly variable and may range from 1 to 12% according to different reports; they may involve both the central and peripheral nervous systems, although the latter is more frequently implicated (central: peripheral = 1:3) [117][118][119]. The challenge for clinicians is that the syndromes associated with ICIs meet diagnostic criteria for PNS, and all alternative etiologies (e.g., carcinomatous meningitis) must be excluded [16]. Recently, consensus guidelines have been developed to appropriately classify n-irAEs [117]. Seven main syndromes have been described, four of which involve the CNS. CNS-irAEs include immune-related (ir) encephalitis, ir meningitis, ir vasculitis, and ir demyelinating diseases [117]. In some cases, n-irAEs may satisfy the clinical diagnostic criteria for PNS associated with “high-risk” antibodies [16][120]. A retrospective study has detected a significant increase of Ma2-associated PNS after the introduction of ICIs in France [121]. There has been substantial interest in discovering biomarkers of disease progression in n-irAEs. For example, patients with ir-encephalitis can show associated antiphosphodiesterase 10A-Abs [122] or an increased absolute eosinophil count [123]. However, these biomarkers and the associated autoantibodies remain of limited clinical applicability. Furthermore, a significant proportion of cases are seronegative despite extensive screening [117][120]. Accordingly, the detection of antibodies is not required for the diagnosis of irAEs. Moreover, although PNS usually precede the discovery of cancer, n-irAEs triggered by ICIs only develop when the cancer is already established and treated, in general, within a short time frame after ICIs have been started.
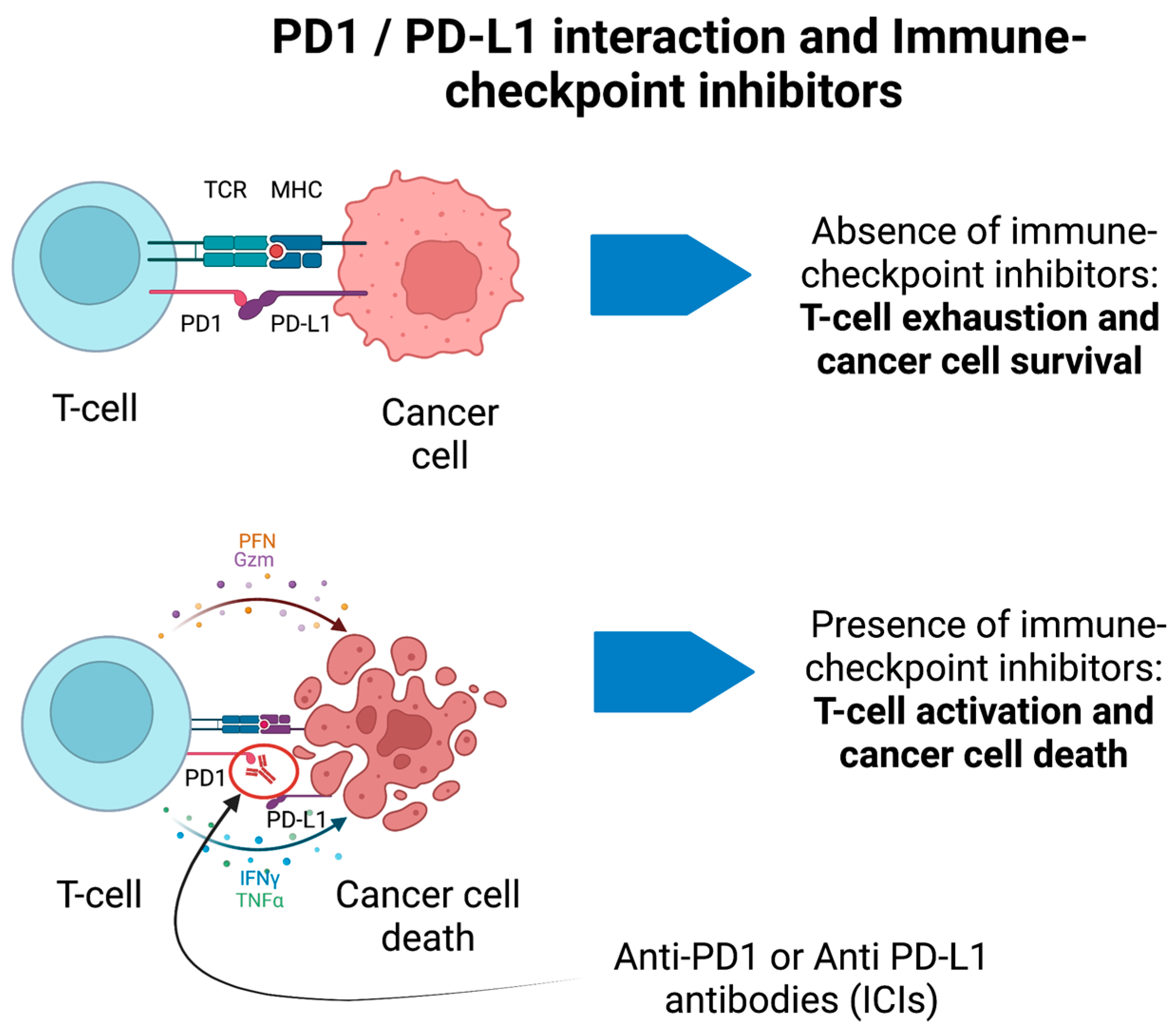
Figure 2. PD1/PD-L1 interaction and immune-checkpoint inhibitors for cancer treatment. In the absence of immune-checkpoint inhibitors, the binding between PD1 and PD-L1 on T-cells and cancer cells, respectively, prevents the activation of T-cells. In presence of anti-PD1 antibodies (ICIs), the T-cell is activated against cancer cells and promotes their death through different immune-mediated pathways. These may generate immune-mediated adverse events.
Therapies based on genetically modified T cells harboring chimeric antigen receptors, also known as CAR T-cell therapies, represent a powerful therapeutic strategy for several hematological cancers and have been associated with significant neurotoxicity [16]. The most aggressive and life-threatening neurotoxicity related to CAR T-cell therapies is the CAR T-cell encephalopathy [124][125]. Forty percent of patients affected by CAR T-cell encephalopathy may have severe or fatal clinical courses [126]. The pathophysiology is thought to arise from the disruption of the blood–brain barrier and the subsequent edema induced by the cytokine release stimulated by CAR T-cell therapies (Figure 3) [124][127]. Symptoms usually follow a stereotyped progression beginning with somnolence, disorientation, confusion, followed by aphasia, hallucinations, and myoclonus. Severe cases progress to generalized seizures and encephalopathy possibly leading to coma and death if not promptly recognized and treated. While not considered part of the PNS, CAR T-cell encephalopathy should be distinguished from other paraneoplastic encephalitides (see Section 4).

Figure 3. CAR T-cell therapies for cancer treatment. CAR T interacts with cancer cell’s antigen (1); it then activates a co-stimulatory signal which stimulates the synthesis, transcription, and translation of perforins and granzymes (2, 3) that ultimately kill the cancer cell (4).
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11051406
References
- Chirra, M.; Marsili, L.; Gallerini, S.; Keeling, E.G.; Marconi, R.; Colosimo, C. Paraneoplastic movement disorders: Phenomenology, diagnosis, and treatment. Eur. J. Intern. Med. 2019, 67, 14–23.
- Dalmau, J.; Rosenfeld, M.R. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008, 7, 327–340.
- Darnell, R.B.; Posner, J.B. Paraneoplastic syndromes involving the nervous system. N. Engl. J. Med. 2003, 349, 1543–1554.
- Pittock, S.J.; Lucchinetti, C.F.; Parisi, J.E.; Benarroch, E.E.; Mokri, B.; Stephan, C.L.; Kim, K.K.; Kilimann, M.W.; Lennon, V.A. Amphiphysin autoimmunity: Paraneoplastic accompaniments. Ann. Neurol. 2005, 58, 96–107.
- Giometto, B.; Grisold, W.; Vitaliani, R.; Graus, F.; Honnorat, J.; Bertolini, G. Paraneoplastic neurologic syndrome in the PNS Euronetwork database: A European study from 20 centers. Arch. Neurol. 2010, 67, 330–335.
- Graus, F.; Delattre, J.Y.; Antoine, J.C.; Dalmau, J.; Giometto, B.; Grisold, W.; Honnorat, J.; Smitt, P.S.; Vedeler, C.; Verschuuren, J.J.; et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1135–1140.
- Chan, A.M.; Baehring, J.M. Paraneoplastic neurological syndromes: A single institution 10-year case series. J. Neurooncol. 2019, 141, 431–439.
- Dubey, D.; Pittock, S.J.; Kelly, C.R.; McKeon, A.; Lopez-Chiriboga, A.S.; Lennon, V.A.; Gadoth, A.; Smith, C.Y.; Bryant, S.C.; Klein, C.J.; et al. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann. Neurol. 2018, 83, 166–177.
- Flanagan, E.P.; McKeon, A.; Lennon, V.A.; Boeve, B.F.; Trenerry, M.R.; Tan, K.M.; Drubach, D.A.; Josephs, K.A.; Britton, J.W.; Mandrekar, J.N.; et al. Autoimmune dementia: Clinical course and predictors of immunotherapy response. Mayo. Clin. Proc. 2010, 85, 881–897.
- Flanagan, E.P.; Geschwind, M.D.; Lopez-Chiriboga, A.S.; Blackburn, K.M.; Turaga, S.; Binks, S.; Zitser, J.; Gelfand, J.M.; Day, G.S.; Dunham, S.R.; et al. Autoimmune Encephalitis Misdiagnosis in Adults. JAMA Neurol. 2023, 80, 30–39.
- Vogrig, A.; Muñiz-Castrillo, S.; Desestret, V.; Joubert, B.; Honnorat, J. Pathophysiology of paraneoplastic and autoimmune encephalitis: Genes, infections, and checkpoint inhibitors. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420932797.
- Honorat, J.A.; Komorowski, L.; Josephs, K.A.; Fechner, K.; St Louis, E.K.; Hinson, S.R.; Lederer, S.; Kumar, N.; Gadoth, A.; Lennon, V.A.; et al. IgLON5 antibody: Neurological accompaniments and outcomes in 20 patients. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e385.
- Storstein, A.; Krossnes, B.K.; Vedeler, C.A. Morphological and immunohistochemical characterization of paraneoplastic cerebellar degeneration associated with Yo antibodies. Acta Neurol. Scand. 2009, 120, 64–67.
- Greenlee, J.E.; Brashear, H.R. Antibodies to cerebellar Purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann. Neurol. 1983, 14, 609–613.
- Vogrig, A.; Bernardini, A.; Gigli, G.L.; Corazza, E.; Marini, A.; Segatti, S.; Fabris, M.; Honnorat, J.; Valente, M. Stroke-Like Presentation of Paraneoplastic Cerebellar Degeneration: A Single-Center Experience and Review of the Literature. Cerebellum 2019, 18, 976–982.
- Graus, F.; Vogrig, A.; Muñiz-Castrillo, S.; Antoine, J.G.; Desestret, V.; Dubey, D.; Giometto, B.; Irani, S.R.; Joubert, B.; Leypoldt, F.; et al. Updated Diagnostic Criteria for Paraneoplastic Neurologic Syndromes. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1014.
- Rodriguez, M.; Truh, L.I.; O’Neill, B.P.; Lennon, V.A. Autoimmune paraneoplastic cerebellar degeneration: Ultrastructural localization of antibody-binding sites in Purkinje cells. Neurology 1988, 38, 1380–1386.
- McKeon, A.; Tracy, J.A.; Pittock, S.J.; Parisi, J.E.; Klein, C.J.; Lennon, V.A. Purkinje cell cytoplasmic autoantibody type 1 accompaniments: The cerebellum and beyond. Arch. Neurol. 2011, 68, 1282–1289.
- Mendes, N.T.; Ronchi, N.R.; Silva, G.D. A Systematic Review on Anti-Yo/PCA-1 Antibody: Beyond Cerebellar Ataxia in Middle-Aged Women with Gynecologic Cancer. Cerebellum 2022.
- Chatham, M.; Niravath, P. Anti-Yo-Associated Paraneoplastic Cerebellar Degeneration: Case Series and Review of Literature. Cureus 2021, 13, e20203.
- de Graaff, E.; Maat, P.; Hulsenboom, E.; van den Berg, R.; van den Bent, M.; Demmers, J.; Lugtenburg, P.J.; Hoogenraad, C.C.; Sillevis Smitt, P. Identification of delta/notch-like epidermal growth factor-related receptor as the Tr antigen in paraneoplastic cerebellar degeneration. Ann. Neurol. 2012, 71, 815–824.
- Bernal, F.; Shams’ili, S.; Rojas, I.; Sanchez-Valle, R.; Saiz, A.; Dalmau, J.; Honnorat, J.; Sillevis Smitt, P.; Graus, F. Anti-Tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin’s disease. Neurology 2003, 60, 230–234.
- Simard, C.; Vogrig, A.; Joubert, B.; Muñiz-Castrillo, S.; Picard, G.; Rogemond, V.; Ducray, F.; Berzero, G.; Psimaras, D.; Antoine, J.C.; et al. Clinical spectrum and diagnostic pitfalls of neurologic syndromes with Ri antibodies. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e699.
- Dubey, D.; Wilson, M.R.; Clarkson, B.; Giannini, C.; Gandhi, M.; Cheville, J.; Lennon, V.A.; Eggers, S.; Devine, M.F.; Mandel-Brehm, C.; et al. Expanded Clinical Phenotype, Oncological Associations, and Immunopathologic Insights of Paraneoplastic Kelch-like Protein-11 Encephalitis. JAMA Neurol. 2020, 77, 1420–1429.
- Maudes, E.; Landa, J.; Muñoz-Lopetegi, A.; Armangue, T.; Alba, M.; Saiz, A.; Graus, F.; Dalmau, J.; Sabater, L. Clinical significance of Kelch-like protein 11 antibodies. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e666.
- Hammami, M.B.; Rezk, M.; Dubey, D. Validation of MATCH score: A predictive tool for identification of patients with kelch-like protein-11 autoantibodies. J. Neurol. Neurosurg. Psychiatry 2023, 94, 171–172.
- Winklehner, M.; Bauer, J.; Endmayr, V.; Schwaiger, C.; Ricken, G.; Motomura, M.; Yoshimura, S.; Shintaku, H.; Ishikawa, K.; Tsuura, Y.; et al. Paraneoplastic Cerebellar Degeneration with P/Q-VGCC vs Yo Autoantibodies. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e200006.
- Titulaer, M.J.; Lang, B.; Verschuuren, J.J. Lambert-Eaton myasthenic syndrome: From clinical characteristics to therapeutic strategies. Lancet Neurol. 2011, 10, 1098–1107.
- Clark, H.B. The Neuropathology of Autoimmune Ataxias. Brain Sci. 2022, 12, 257.
- Taraghikhah, N.; Ashtari, S.; Asri, N.; Shahbazkhani, B.; Al-Dulaimi, D.; Rostami-Nejad, M.; Rezaei-Tavirani, M.; Razzaghi, M.R.; Zali, M.R. An updated overview of spectrum of gluten-related disorders: Clinical and diagnostic aspects. BMC Gastroenterol. 2020, 20, 258.
- Thapa, S.; Shah, S.; Chand, S.; Sah, S.K.; Gyawali, P.; Paudel, S.; Khanal, P. Ataxia due to vitamin E deficiency: A case report and updated review. Clin. Case Rep. 2022, 10, e6303.
- Grahn, A.; Studahl, M. Varicella-zoster virus infections of the central nervous system—Prognosis, diagnostics and treatment. J. Infect. 2015, 71, 281–293.
- Roy, U.; Panwar, A.; Pandit, A.; Das, S.K.; Joshi, B. Clinical and Neuroradiological Spectrum of Metronidazole Induced Encephalopathy: Our Experience and the Review of Literature. J. Clin. Diagn. Res. 2016, 10, Oe01–Oe09.
- Liampas, A.; Nteveros, A.; Parperis, K.; Akil, M.; Dardiotis, E.; Andreadou, E.; Hadjivassiliou, M.; Zis, P. Primary Sjögren’s syndrome (pSS)-related cerebellar ataxia: A systematic review and meta-analysis. Acta Neurol. Belg. 2022, 122, 457–463.
- Manto, M.; Hadjivassiliou, M.; Baizabal-Carvallo, J.F.; Hampe, C.S.; Honnorat, J.; Joubert, B.; Mitoma, H.; Muñiz-Castrillo, S.; Shaikh, A.G.; Vogrig, A. Consensus Paper: Latent Autoimmune Cerebellar Ataxia (LACA). Cerebellum 2023, 1–18.
- Gallerini, S.; Marsili, L.; Marconi, R. Opsoclonus-Myoclonus Syndrome in the Era of Neuronal Cell Surface Antibodies: A Message for Clinicians. JAMA Neurol. 2016, 73, 891.
- Armangué, T.; Sabater, L.; Torres-Vega, E.; Martínez-Hernández, E.; Ariño, H.; Petit-Pedrol, M.; Planagumà, J.; Bataller, L.; Dalmau, J.; Graus, F. Clinical and Immunological Features of Opsoclonus-Myoclonus Syndrome in the Era of Neuronal Cell Surface Antibodies. JAMA Neurol. 2016, 73, 417–424.
- Graus, F.; Ariño, H.; Dalmau, J. Opsoclonus-Myoclonus Syndrome in the Era of Neuronal Cell Surface Antibodies-Reply. JAMA Neurol. 2016, 73, 891.
- Oh, S.Y.; Kim, J.S.; Dieterich, M. Update on opsoclonus-myoclonus syndrome in adults. J. Neurol. 2019, 266, 1541–1548.
- Krug, P.; Schleiermacher, G.; Michon, J.; Valteau-Couanet, D.; Brisse, H.; Peuchmaur, M.; Sarnacki, S.; Martelli, H.; Desguerre, I.; Tardieu, M. Opsoclonus-myoclonus in children associated or not with neuroblastoma. Eur. J. Paediatr. Neurol. 2010, 14, 400–409.
- Gallerini, S.; Marsili, L. Pediatric opsoclonus-myoclonus syndrome: The role of functional brain connectivity studies. Dev. Med. Child Neurol. 2017, 59, 14–15.
- Bhatia, P.; Heim, J.; Cornejo, P.; Kane, L.; Santiago, J.; Kruer, M.C. Opsoclonus-myoclonus-ataxia syndrome in children. J. Neurol. 2022, 269, 750–757.
- Dale, R.C. Childhood opsoclonus myoclonus. Lancet Neurol. 2003, 2, 270.
- Du, H.; Cai, W. Opsoclonus-myoclonus syndrome associated with neuroblastoma: Insights into antitumor immunity. Pediatr. Blood Cancer 2022, 69, e29949.
- Emamikhah, M.; Babadi, M.; Mehrabani, M.; Jalili, M.; Pouranian, M.; Daraie, P.; Mohaghegh, F.; Aghavali, S.; Zaribafian, M.; Rohani, M. Opsoclonus-myoclonus syndrome, a post-infectious neurologic complication of COVID-19: Case series and review of literature. J. Neurovirol. 2021, 27, 26–34.
- Santoro, J.D.; Kerr, L.M.; Codden, R.; Casper, T.C.; Greenberg, B.M.; Waubant, E.; Kong, S.W.; Mandl, K.D.; Gorman, M.P. Increased Prevalence of Familial Autoimmune Disease in Children With Opsoclonus-Myoclonus Syndrome. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1079.
- Alburaiky, S.; Dale, R.C.; Crow, Y.J.; Jones, H.F.; Wassmer, E.; Melki, I.; Boespflug-Tanguy, O.; Do Cao, J.; Gras, D.; Sharpe, C. Opsoclonus-myoclonus in Aicardi-Goutières syndrome. Dev. Med. Child Neurol. 2021, 63, 1483–1486.
- Graus, F. Towards a better recognition of paraneoplastic brainstem encephalitis. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1141.
- Kim, K.T.; Baek, S.H.; Lee, S.U.; Kim, J.B.; Kim, J.S. Clinical Reasoning: A 48-Year-Old Woman Presenting With Vertigo, Ptosis, and Red Eyes. Neurology 2022, 98, 678–683.
- Sutton, I.J.; Barnett, M.H.; Watson, J.D.; Ell, J.J.; Dalmau, J. Paraneoplastic brainstem encephalitis and anti-Ri antibodies. J. Neurol. 2002, 249, 1597–1598.
- Ohyagi, M.; Ishibashi, S.; Ohkubo, T.; Kobayashi, Z.; Mizusawa, H.; Yokota, T.; Emoto, H.; Kiyosawa, M. Subacute Supranuclear Palsy in anti-Hu Paraneoplastic Encephalitis. Can. J. Neurol. Sci. 2017, 44, 444–446.
- Najjar, M.; Taylor, A.; Agrawal, S.; Fojo, T.; Merkler, A.E.; Rosenblum, M.K.; Lennihan, L.; Kluger, M.D. Anti-Hu paraneoplastic brainstem encephalitis caused by a pancreatic neuroendocrine tumor presenting with central hypoventilation. J. Clin. Neurosci. 2017, 40, 72–73.
- Berger, B.; Bischler, P.; Dersch, R.; Hottenrott, T.; Rauer, S.; Stich, O. “Non-classical” paraneoplastic neurological syndromes associated with well-characterized antineuronal antibodies as compared to “classical” syndromes—More frequent than expected. J. Neurol. Sci. 2015, 352, 58–61.
- Mandel-Brehm, C.; Dubey, D.; Kryzer, T.J.; O’Donovan, B.D.; Tran, B.; Vazquez, S.E.; Sample, H.A.; Zorn, K.C.; Khan, L.M.; Bledsoe, I.O.; et al. Kelch-like Protein 11 Antibodies in Seminoma-Associated Paraneoplastic Encephalitis. N. Engl. J. Med. 2019, 381, 47–54.
- Di Schino, C.; Nunzi, M.; Colosimo, C. Subacute axial parkinsonism associated with anti-Ri antibodies. Neurol. Sci. 2021, 42, 1155–1156.
- Dalmau, J.; Graus, F.; Villarejo, A.; Posner, J.B.; Blumenthal, D.; Thiessen, B.; Saiz, A.; Meneses, P.; Rosenfeld, M.R. Clinical analysis of anti-Ma2-associated encephalitis. Brain 2004, 127, 1831–1844.
- Yamamoto, T.; Tsuji, S. Anti-Ma2-associated encephalitis and paraneoplastic limbic encephalitis. Brain Nerve 2010, 62, 838–851.
- Adams, C.; McKeon, A.; Silber, M.H.; Kumar, R. Narcolepsy, REM sleep behavior disorder, and supranuclear gaze palsy associated with Ma1 and Ma2 antibodies and tonsillar carcinoma. Arch. Neurol. 2011, 68, 521–524.
- Xing, F.; Marsili, L.; Truong, D.D. Parkinsonism in Viral, Paraneoplastic, and Autoimmune Diseases. J. Neurol. Sci. 2022, 433, e120014.
- Vogrig, A.; Joubert, B.; Maureille, A.; Thomas, L.; Bernard, E.; Streichenberger, N.; Cotton, F.; Ducray, F.; Honnorat, J. Motor neuron involvement in anti-Ma2-associated paraneoplastic neurological syndrome. J. Neurol. 2019, 266, 398–410.
- Tomar, L.R.; Agarwal, U.; Shah, D.J.; Jain, S.; Agrawal, C.S. Jaw Dystonia and Myelopathy: Paraneoplastic Manifestations of Breast Malignancy with anti-Ri/ANNA-2 Antibody. Ann. Indian Acad. Neurol. 2021, 24, 826–828.
- Ortega Suero, G.; Sola-Valls, N.; Escudero, D.; Saiz, A.; Graus, F. Anti-Ma and anti-Ma2-associated paraneoplastic neurological syndromes. Neurologia 2018, 33, 18–27.
- Kunchok, A.; McKeon, A. Opsoclonus in Anti-Ma2 Brain-Stem Encephalitis. N. Engl. J. Med. 2020, 383, e84.
- Orozco, E.; Valencia-Sanchez, C.; Britton, J.; Dubey, D.; Flanagan, E.P.; Lopez-Chiriboga, A.S.; Zalewski, N.; Zekeridou, A.; Pittock, S.J.; McKeon, A. Autoimmune Encephalitis Criteria in Clinical Practice. Neurol. Clin. Pract. 2023, 13, e200151.
- Ghimire, P.; Khanal, U.P.; Gajurel, B.P.; Karn, R.; Rajbhandari, R.; Paudel, S.; Gautam, N.; Ojha, R. Anti-LGI1, anti-GABABR, and Anti-CASPR2 encephalitides in Asia: A systematic review. Brain Behav. 2020, 10, e01793.
- Alamowitch, S.; Graus, F.; Uchuya, M.; Reñé, R.; Bescansa, E.; Delattre, J.Y. Limbic encephalitis and small cell lung cancer. Clinical and immunological features. Brain 1997, 120 Pt 6, 923–928.
- Graus, F.; Keime-Guibert, F.; Reñe, R.; Benyahia, B.; Ribalta, T.; Ascaso, C.; Escaramis, G.; Delattre, J.Y. Anti-Hu-associated paraneoplastic encephalomyelitis: Analysis of 200 patients. Brain 2001, 124, 1138–1148.
- Steriade, C.; Britton, J.; Dale, R.C.; Gadoth, A.; Irani, S.R.; Linnoila, J.; McKeon, A.; Shao, X.Q.; Venegas, V.; Bien, C.G. Acute symptomatic seizures secondary to autoimmune encephalitis and autoimmune-associated epilepsy: Conceptual definitions. Epilepsia 2020, 61, 1341–1351.
- Shin, K.J.; Ji, Y.I. Anti-Hu antibody-mediated limbic encephalitis associated with cervical cancer: A case report. J. Obstet. Gynaecol. Res. 2018, 44, 1181–1184.
- Silsby, M.; Clarke, C.J.; Lee, K.; Sharpe, D. Anti-Hu limbic encephalitis preceding the appearance of mediastinal germinoma by 9 years. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e685.
- Honnorat, J.; Didelot, A.; Karantoni, E.; Ville, D.; Ducray, F.; Lambert, L.; Deiva, K.; Garcia, M.; Pichit, P.; Cavillon, G.; et al. Autoimmune limbic encephalopathy and anti-Hu antibodies in children without cancer. Neurology 2013, 80, 2226–2232.
- Dalmau, J.; Tüzün, E.; Wu, H.Y.; Masjuan, J.; Rossi, J.E.; Voloschin, A.; Baehring, J.M.; Shimazaki, H.; Koide, R.; King, D.; et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann. Neurol. 2007, 61, 25–36.
- Titulaer, M.J.; McCracken, L.; Gabilondo, I.; Armangué, T.; Glaser, C.; Iizuka, T.; Honig, L.S.; Benseler, S.M.; Kawachi, I.; Martinez-Hernandez, E.; et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: An observational cohort study. Lancet Neurol. 2013, 12, 157–165.
- Bost, C.; Chanson, E.; Picard, G.; Meyronet, D.; Mayeur, M.E.; Ducray, F.; Rogemond, V.; Psimaras, D.; Antoine, J.C.; Delattre, J.Y.; et al. Malignant tumors in autoimmune encephalitis with anti-NMDA receptor antibodies. J. Neurol. 2018, 265, 2190–2200.
- Chakraborty, A.P.; Pandit, A.; Ray, B.K.; Mukherjee, A.; Dubey, S. Capgras syndrome and confabulation unfurling anti NMDAR encephalitis with classical papillary thyroid carcinoma: First reported case. J. Neuroimmunol. 2021, 357, 577611.
- Yang, J.; Li, B.; Li, X.; Lai, Z. Anti-N-Methyl-D-Aspartate Receptor Encephalitis Associated with Clear Cell Renal Carcinoma: A Case Report. Front. Oncol. 2020, 10, 350.
- Shalhout, S.Z.; Emerick, K.S.; Sadow, P.M.; Linnoila, J.J.; Miller, D.M. Regionally Metastatic Merkel Cell Carcinoma Associated with Paraneoplastic Anti-N-methyl-D-aspartate Receptor Encephalitis. Case Rep. Oncol. Med. 2020, 2020, 1257587.
- Prüss, H.; Finke, C.; Höltje, M.; Hofmann, J.; Klingbeil, C.; Probst, C.; Borowski, K.; Ahnert-Hilger, G.; Harms, L.; Schwab, J.M.; et al. N-methyl-D-aspartate receptor antibodies in herpes simplex encephalitis. Ann. Neurol. 2012, 72, 902–911.
- Leypoldt, F.; Titulaer, M.J.; Aguilar, E.; Walther, J.; Bönstrup, M.; Havemeister, S.; Teegen, B.; Lütgehetmann, M.; Rosenkranz, M.; Magnus, T.; et al. Herpes simplex virus-1 encephalitis can trigger anti-NMDA receptor encephalitis: Case report. Neurology 2013, 81, 1637–1639.
- Salovin, A.; Glanzman, J.; Roslin, K.; Armangue, T.; Lynch, D.R.; Panzer, J.A. Anti-NMDA receptor encephalitis and nonencephalitic HSV-1 infection. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e458.
- Hu, S.; Lan, T.; Bai, R.; Jiang, S.; Cai, J.; Ren, L. HSV encephalitis triggered anti-NMDAR encephalitis: A case report. Neurol. Sci. 2021, 42, 857–861.
- Liu, X.; Yan, B.; Wang, R.; Li, C.; Chen, C.; Zhou, D.; Hong, Z. Seizure outcomes in patients with anti-NMDAR encephalitis: A follow-up study. Epilepsia 2017, 58, 2104–2111.
- Schmitt, S.E.; Pargeon, K.; Frechette, E.S.; Hirsch, L.J.; Dalmau, J.; Friedman, D. Extreme delta brush: A unique EEG pattern in adults with anti-NMDA receptor encephalitis. Neurology 2012, 79, 1094–1100.
- Moise, A.M.; Karakis, I.; Herlopian, A.; Dhakar, M.; Hirsch, L.J.; Cotsonis, G.; LaRoche, S.; Cabrera Kang, C.M.; Westover, B.; Rodriguez, A. Continuous EEG Findings in Autoimmune Encephalitis. J. Clin. Neurophysiol. 2021, 38, 124–129.
- Dalmau, J.; Armangué, T.; Planagumà, J.; Radosevic, M.; Mannara, F.; Leypoldt, F.; Geis, C.; Lancaster, E.; Titulaer, M.J.; Rosenfeld, M.R.; et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: Mechanisms and models. Lancet Neurol. 2019, 18, 1045–1057.
- Irani, S.R.; Alexander, S.; Waters, P.; Kleopa, K.A.; Pettingill, P.; Zuliani, L.; Peles, E.; Buckley, C.; Lang, B.; Vincent, A. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010, 133, 2734–2748.
- Irani, S.R.; Pettingill, P.; Kleopa, K.A.; Schiza, N.; Waters, P.; Mazia, C.; Zuliani, L.; Watanabe, O.; Lang, B.; Buckley, C.; et al. Morvan syndrome: Clinical and serological observations in 29 cases. Ann. Neurol. 2012, 72, 241–255.
- Benoit, J.; Muñiz-Castrillo, S.; Vogrig, A.; Farina, A.; Pinto, A.L.; Picard, G.; Rogemond, V.; Guery, D.; Alentorn, A.; Psimaras, D.; et al. Early-Stage Contactin-Associated Protein-like 2 Limbic Encephalitis: Clues for Diagnosis. Neurol. Neuroimmunol. Neuroinflamm. 2023, 10, e200041.
- van Sonderen, A.; Thijs, R.D.; Coenders, E.C.; Jiskoot, L.C.; Sanchez, E.; de Bruijn, M.A.; van Coevorden-Hameete, M.H.; Wirtz, P.W.; Schreurs, M.W.; Sillevis Smitt, P.A.; et al. Anti-LGI1 encephalitis: Clinical syndrome and long-term follow-up. Neurology 2016, 87, 1449–1456.
- Griffith, S.P.; Malpas, C.B.; Alpitsis, R.; O’Brien, T.J.; Monif, M. The neuropsychological spectrum of anti-LGI1 antibody mediated autoimmune encephalitis. J. Neuroimmunol. 2020, 345, 577271.
- Rodriguez, A.; Klein, C.J.; Sechi, E.; Alden, E.; Basso, M.R.; Pudumjee, S.; Pittock, S.J.; McKeon, A.; Britton, J.W.; Lopez-Chiriboga, A.S.; et al. LGI1 antibody encephalitis: Acute treatment comparisons and outcome. J. Neurol. Neurosurg. Psychiatry. 2022, 93, 309–315.
- Lin, N.; Hao, H.; Guan, H.; Sun, H.; Liu, Q.; Lu, Q.; Jin, L.; Ren, H.; Huang, Y. Sleep Disorders in Leucine-Rich Glioma-Inactivated Protein 1 and Contactin Protein-Like 2 Antibody-Associated Diseases. Front. Neurol. 2020, 11, 696.
- Piffer, S.; Cantalupo, G.; Filipponi, S.; Poretto, V.; Pellegrini, M.; Tanel, R.; Buganza, M.; Giometto, B. Agrypnia excitata as the main feature in anti-leucine-rich glioma-inactivated 1 encephalitis: A detailed clinical and polysomnographic semiological analysis. Eur. J. Neurol. 2022, 29, 890–894.
- Baldelli, L.; Provini, F. Differentiating Oneiric Stupor in Agrypnia Excitata From Dreaming Disorders. Front. Neurol. 2020, 11, 565694.
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, R.C.; Gelfand, J.M.; Geschwind, M.; et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016, 15, 391–404.
- Jia, Y.; Wang, H.; Zhang, M.; Wei, M.; Huang, Z.; Ye, J.; Liu, A.; Wang, Y. LGI1 antibody-associated encephalitis without evidence of inflammation in CSF and brain MRI. Acta Neurol. Belg. 2022.
- Abgrall, G.; Demeret, S.; Rohaut, B.; Leu-Semenescu, S.; Arnulf, I. Status dissociatus and disturbed dreaming in a patient with Morvan syndrome plus myasthenia gravis. Sleep Med. 2015, 16, 894–896.
- Nagappa, M.; Mahadevan, A.; Sinha, S.; Bindu, P.S.; Mathuranath, P.S.; Bineesh, C.; Bharath, R.D.; Taly, A.B. Fatal Morvan Syndrome Associated With Myasthenia Gravis. Neurologist 2017, 22, 29–33.
- Boyko, M.; Au, K.L.K.; Casault, C.; de Robles, P.; Pfeffer, G. Systematic review of the clinical spectrum of CASPR2 antibody syndrome. J. Neurol. 2020, 267, 1137–1146.
- Lancaster, E.; Lai, M.; Peng, X.; Hughes, E.; Constantinescu, R.; Raizer, J.; Friedman, D.; Skeen, M.B.; Grisold, W.; Kimura, A.; et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: Case series and characterisation of the antigen. Lancet Neurol. 2010, 9, 67–76.
- Höftberger, R.; Titulaer, M.J.; Sabater, L.; Dome, B.; Rózsás, A.; Hegedus, B.; Hoda, M.A.; Laszlo, V.; Ankersmit, H.J.; Harms, L.; et al. Encephalitis and GABAB receptor antibodies: Novel findings in a new case series of 20 patients. Neurology 2013, 81, 1500–1506.
- de Bruijn, M.; van Sonderen, A.; van Coevorden-Hameete, M.H.; Bastiaansen, A.E.M.; Schreurs, M.W.J.; Rouhl, R.P.W.; van Donselaar, C.A.; Majoie, M.; Neuteboom, R.F.; Sillevis Smitt, P.A.E.; et al. Evaluation of seizure treatment in anti-LGI1, anti-NMDAR, and anti-GABA(B)R encephalitis. Neurology 2019, 92, e2185–e2196.
- Zhang, Z.; Fan, S.; Ren, H.; Zhou, L.; Guan, H. Clinical characteristics and prognosis of anti-alpha-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic acid receptor encephalitis. BMC Neurol. 2021, 21, 490.
- Joubert, B.; Kerschen, P.; Zekeridou, A.; Desestret, V.; Rogemond, V.; Chaffois, M.O.; Ducray, F.; Larrue, V.; Daubail, B.; Idbaih, A.; et al. Clinical Spectrum of Encephalitis Associated With Antibodies Against the α-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid Receptor: Case Series and Review of the Literature. JAMA Neurol. 2015, 72, 1163–1169.
- Höftberger, R.; van Sonderen, A.; Leypoldt, F.; Houghton, D.; Geschwind, M.; Gelfand, J.; Paredes, M.; Sabater, L.; Saiz, A.; Titulaer, M.J.; et al. Encephalitis and AMPA receptor antibodies: Novel findings in a case series of 22 patients. Neurology 2015, 84, 2403–2412.
- McKeon, A.; Robinson, M.T.; McEvoy, K.M.; Matsumoto, J.Y.; Lennon, V.A.; Ahlskog, J.E.; Pittock, S.J. Stiff-man syndrome and variants: Clinical course, treatments, and outcomes. Arch. Neurol. 2012, 69, 230–238.
- Martinez-Hernandez, E.; Ariño, H.; McKeon, A.; Iizuka, T.; Titulaer, M.J.; Simabukuro, M.M.; Lancaster, E.; Petit-Pedrol, M.; Planagumà, J.; Blanco, Y.; et al. Clinical and Immunologic Investigations in Patients With Stiff-Person Spectrum Disorder. JAMA Neurol. 2016, 73, 714–720.
- Budhram, A.; Sechi, E.; Flanagan, E.P.; Dubey, D.; Zekeridou, A.; Shah, S.S.; Gadoth, A.; Naddaf, E.; McKeon, A.; Pittock, S.J.; et al. Clinical spectrum of high-titre GAD65 antibodies. J. Neurol. Neurosurg. Psychiatry 2021, 92, 645–654.
- Sasaki, A.; Kato, T.; Ujiie, H.; Wakasa, S.; Otake, S.; Kikuchi, K.; Ohno, K. Thymoma-Related Stiff-Person Syndrome with Successfully Treated by Surgery. Ann. Thorac. Cardiovasc. Surg. 2022, 28, 448–452.
- Murinson, B.B.; Guarnaccia, J.B. Stiff-person syndrome with amphiphysin antibodies: Distinctive features of a rare disease. Neurology 2008, 71, 1955–1958.
- McKeon, A.; Pittock, S.J.; Lennon, V.A. Stiff-person syndrome with amphiphysin antibodies: Distinctive features of a rare disease. Neurology 2009, 73, 2132–2133.
- Carvajal-González, A.; Leite, M.I.; Waters, P.; Woodhall, M.; Coutinho, E.; Balint, B.; Lang, B.; Pettingill, P.; Carr, A.; Sheerin, U.M.; et al. Glycine receptor antibodies in PERM and related syndromes: Characteristics, clinical features and outcomes. Brain 2014, 137, 2178–2192.
- Balint, B.; Jarius, S.; Nagel, S.; Haberkorn, U.; Probst, C.; Blöcker, I.M.; Bahtz, R.; Komorowski, L.; Stöcker, W.; Kastrup, A.; et al. Progressive encephalomyelitis with rigidity and myoclonus: A new variant with DPPX antibodies. Neurology 2014, 82, 1521–1528.
- Tobin, W.O.; Lennon, V.A.; Komorowski, L.; Probst, C.; Clardy, S.L.; Aksamit, A.J.; Appendino, J.P.; Lucchinetti, C.F.; Matsumoto, J.Y.; Pittock, S.J.; et al. DPPX potassium channel antibody: Frequency, clinical accompaniments, and outcomes in 20 patients. Neurology 2014, 83, 1797–1803.
- McKeon, A.; Martinez-Hernandez, E.; Lancaster, E.; Matsumoto, J.Y.; Harvey, R.J.; McEvoy, K.M.; Pittock, S.J.; Lennon, V.A.; Dalmau, J. Glycine receptor autoimmune spectrum with stiff-man syndrome phenotype. JAMA Neurol. 2013, 70, 44–50.
- Huang, P.W.; Chang, J.W. Immune checkpoint inhibitors win the 2018 Nobel Prize. Biomed. J. 2019, 42, 299–306.
- Guidon, A.C.; Burton, L.B.; Chwalisz, B.K.; Hillis, J.; Schaller, T.H.; Amato, A.A.; Betof Warner, A.; Brastianos, P.K.; Cho, T.A.; Clardy, S.L.; et al. Consensus disease definitions for neurologic immune-related adverse events of immune checkpoint inhibitors. J. Immunother. Cancer 2021, 9, e002890.
- Marini, A.; Bernardini, A.; Gigli, G.L.; Valente, M.; Muñiz-Castrillo, S.; Honnorat, J.; Vogrig, A. Neurologic Adverse Events of Immune Checkpoint Inhibitors: A Systematic Review. Neurology 2021, 96, 754–766.
- Marsili, L.; Vogrig, A.; Colosimo, C. Movement Disorders in Oncology: From Clinical Features to Biomarkers. Biomedicines 2021, 10, 26.
- Graus, F.; Dalmau, J. Paraneoplastic neurological syndromes in the era of immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2019, 16, 535–548.
- Vogrig, A.; Fouret, M.; Joubert, B.; Picard, G.; Rogemond, V.; Pinto, A.L.; Muñiz-Castrillo, S.; Roger, M.; Raimbourg, J.; Dayen, C.; et al. Increased frequency of anti-Ma2 encephalitis associated with immune checkpoint inhibitors. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e604.
- Zekeridou, A.; Kryzer, T.; Guo, Y.; Hassan, A.; Lennon, V.; Lucchinetti, C.F.; Pittock, S.; McKeon, A. Phosphodiesterase 10A IgG: A novel biomarker of paraneoplastic neurologic autoimmunity. Neurology 2019, 93, e815–e822.
- Giommoni, E.; Giorgione, R.; Paderi, A.; Pellegrini, E.; Gambale, E.; Marini, A.; Antonuzzo, A.; Marconcini, R.; Roviello, G.; Matucci-Cerinic, M.; et al. Eosinophil Count as Predictive Biomarker of Immune-Related Adverse Events (irAEs) in Immune Checkpoint Inhibitors (ICIs) Therapies in Oncological Patients. Immuno 2021, 1, 253–263.
- Perrinjaquet, C.; Desbaillets, N.; Hottinger, A.F. Neurotoxicity associated with cancer immunotherapy: Immune checkpoint inhibitors and chimeric antigen receptor T-cell therapy. Curr. Opin. Neurol. 2019, 32, 500–510.
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971.
- Torre, M.; Solomon, I.H.; Sutherland, C.L.; Nikiforow, S.; DeAngelo, D.J.; Stone, R.M.; Vaitkevicius, H.; Galinsky, I.A.; Padera, R.F.; Trede, N.; et al. Neuropathology of a Case With Fatal CAR T-Cell-Associated Cerebral Edema. J. Neuropathol. Exp. Neurol. 2018, 77, 877–882.
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62.
This entry is offline, you can click here to edit this entry!