The tumor microenvironment regulates many aspects of cancer progression and anti-tumor immunity. Cancer cells employ a variety of immunosuppressive mechanisms to dampen immune cell function in the tumor microenvironment. While immunotherapies that target these mechanisms, such as immune checkpoint blockade, have had notable clinical success, resistance is common, and there is an urgent need to identify additional targets. Extracellular adenosine, a metabolite of ATP, is found at high levels in the tumor microenvironment and has potent immunosuppressive properties. Targeting members of the adenosine signaling pathway represents a promising immunotherapeutic modality that can potentially synergize with conventional anti-cancer treatment strategies.
- adenosine
- immunotherapy
- treatment combination
1. Introduction
2. Adenosine-Pathway-Mediated Immunosuppression
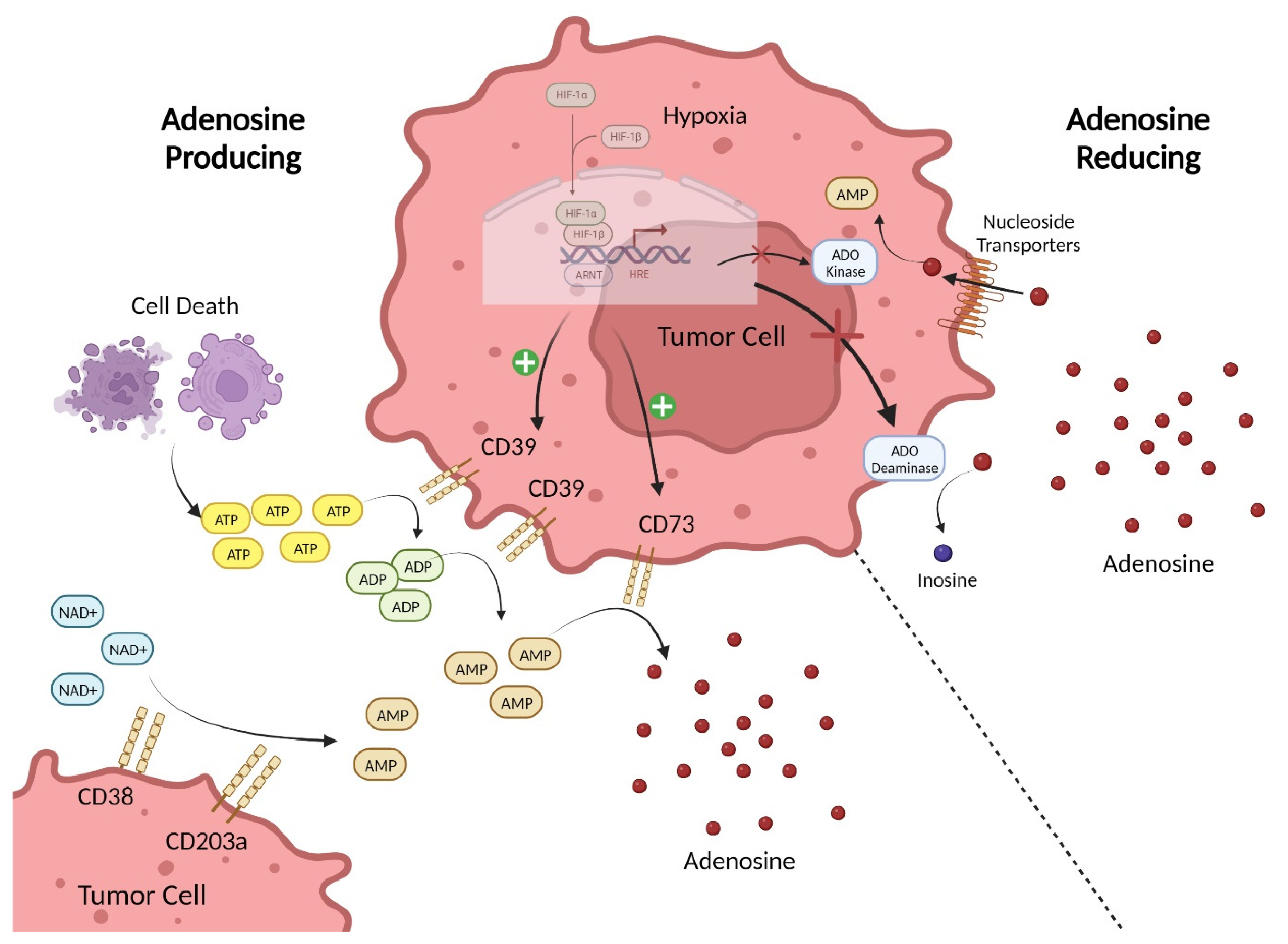
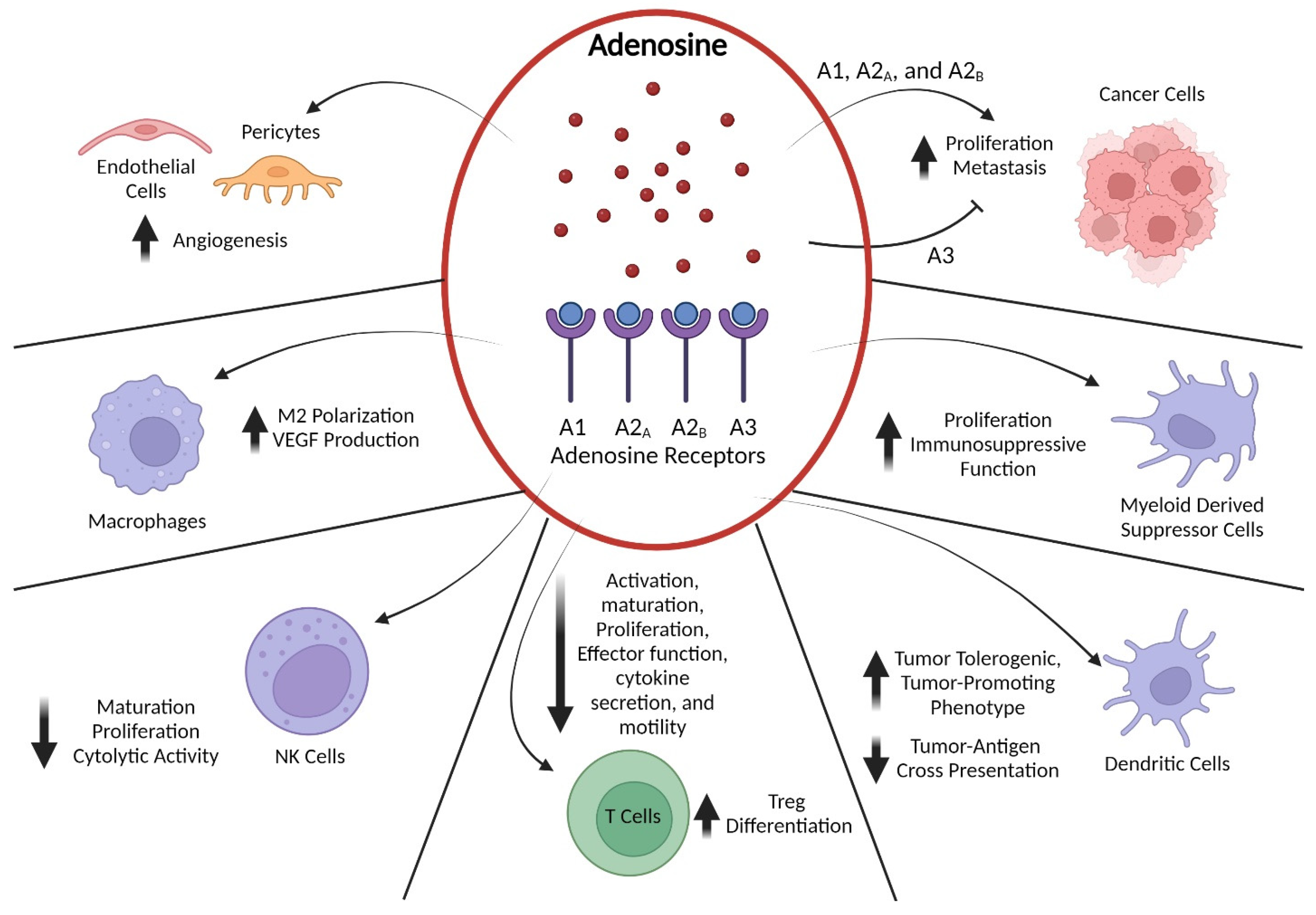
3. Preclinical and Clinical Data Supporting Adenosine-Pathway-Targeted Therapies
4. Potential Combinations with Adenosine-Pathway-Targeted Therapy
This entry is adapted from the peer-reviewed paper 10.3390/ijms24108871
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312.
- Tormoen, G.W.; Crittenden, M.R.; Gough, M.J. Role of the immunosuppressive microenvironment in immunotherapy. Adv. Radiat. Oncol. 2018, 3, 520–526.
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274.
- Tuccitto, A.; Shahaj, E.; Vergani, E.; Ferro, S.; Huber, V.; Rodolfo, M.; Castelli, C.; Rivoltini, L.; Vallacchi, V. Immunosuppressive circuits in tumor microenvironment and their influence on cancer treatment efficacy. Virchows Arch. 2018, 474, 407–420.
- Feng, L.-L.; Cai, Y.-Q.; Zhu, M.-C.; Xing, L.-J.; Wang, X. The yin and yang functions of extracellular ATP and adenosine in tumor immunity. Cancer Cell Int. 2020, 20, 110–111.
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618.
- Sek, K.; Mølck, C.; Stewart, G.D.; Kats, L.; Darcy, P.K.; Beavis, P.A. Targeting Adenosine Receptor Signaling in Cancer Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3837.
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31.
- Li, Y.; Huang, H.; Bin Liu, B.; Zhang, Y.; Pan, X.; Yu, X.-Y.; Shen, Z.; Song, Y.-H. Inflammasomes as therapeutic targets in human diseases. Signal Transduct. Target. Ther. 2021, 6, 247.
- Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Bhol, C.S.; Praharaj, P.P.; Panigrahi, D.P.; Patra, S.; Singh, A.; Patil, S.; Dhiman, R.; et al. Inflammasomes in cancer: Effect of epigenetic and autophagic modulations. Semin. Cancer Biol. 2022, 83, 399–412.
- Lin, T.-Y.; Tsai, M.-C.; Tu, W.; Yeh, H.-C.; Wang, S.-C.; Huang, S.-P.; Li, C.-Y. Role of the NLRP3 Inflammasome: Insights into Cancer Hallmarks. Front. Immunol. 2021, 11, 610492.
- Stagg, J.; Smyth, M.J. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 2010, 29, 5346–5358.
- Long, J.S.; Crighton, D.; O’prey, J.; MacKay, G.; Zheng, L.; Palmer, T.M.; Gottlieb, E.; Ryan, K.M. Extracellular Adenosine Sensing—A Metabolic Cell Death Priming Mechanism Downstream of p53. Mol. Cell 2013, 50, 394–406.
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925.
- Xia, C.; Yin, S.; To, K.K.W.; Fu, L. CD39/CD73/A2AR pathway and cancer immunotherapy. Mol. Cancer 2023, 22, 44.
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144.
- Horenstein, A.L.; Quarona, V.; Toscani, D.; Costa, F.; Chillemi, A.; Pistoia, V.; Giuliani, N.; Malavasi, F. Adenosine Generated in the Bone Marrow Niche Through a CD38-Mediated Pathway Correlates with Progression of Human Myeloma. Mol. Med. 2016, 22, 694–704.
- Linden, J. Adenosine metabolism and cancer. Focus on “Adenosine downregulates DPPIV on HT-29 colon cancer cells by stimulating protein tyrosine phosphatases and reducing ERK1/2 activity via a novel pathway”. Am. J. Physiol. Physiol. 2006, 291, C405–C406.
- Chang, C.-P.; Wu, K.-C.; Lin, C.-Y.; Chern, Y. Emerging roles of dysregulated adenosine homeostasis in brain disorders with a specific focus on neurodegenerative diseases. J. Biomed. Sci. 2021, 28, 70.
- Morote-Garcia, J.C.; Rosenberger, P.; Kuhlicke, J.; Eltzschig, H.K. HIF-1–dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood 2008, 111, 5571–5580.
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629.
- Sheth, S.; Brito, R.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Adenosine Receptors: Expression, Function and Regulation. Int. J. Mol. Sci. 2014, 15, 2024–2052.
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625.
- Borea, P.A.; Varani, K.; Gessi, S.; Merighi, S.; Vincenzi, F. (Eds.) The Adenosine Receptors; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; Volume 34.
- Cekic, C.; Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177–192.
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801.
- Bahreyni, A.; Samani, S.S.; Rahmani, F.; Behnam-Rassouli, R.; Khazaei, M.; Ryzhikov, M.; Parizadeh, M.R.; Avan, A.; Hassanian, S.M. Role of adenosine signaling in the pathogenesis of breast cancer. J. Cell. Physiol. 2017, 233, 1836–1843.
- Khayami, R.; Toroghian, Y.; Bahreyni, A.; Bahrami, A.; Khazaei, M.; Ferns, G.A.; Ebrahimi, S.; Soleimani, A.; Fiuji, H.; Avan, A.; et al. Role of adenosine signaling in the pathogenesis of head and neck cancer. J. Cell. Biochem. 2018, 119, 7905–7912.
- Bova, V.; Filippone, A.; Casili, G.; Lanza, M.; Campolo, M.; Capra, A.P.; Repici, A.; Crupi, L.; Motta, G.; Colarossi, C.; et al. Adenosine Targeting as a New Strategy to Decrease Glioblastoma Aggressiveness. Cancers 2022, 14, 4032.
- Hajizadeh, F.; Masjedi, A.; Asl, S.H.; Kiani, F.K.; Peydaveisi, M.; Ghalamfarsa, G.; Jadidi-Niaragh, F.; Sevbitov, A. Adenosine and adenosine receptors in colorectal cancer. Int. Immunopharmacol. 2020, 87, 106853.
- Wang, J.; Du, L.; Chen, X. Adenosine signaling: Optimal target for gastric cancer immunotherapy. Front. Immunol. 2022, 13, 1027838.
- Wolberg, G.; Zimmerman, T.P.; Hiemstra, K.; Winston, M.; Chu, L.-C. Adenosine Inhibition of Lymphocyte-Mediated Cytolysis: Possible Role of Cyclic Adenosine Monophosphate. Science 1975, 187, 957–959.
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137.
- Waickman, A.T.; Alme, A.; Senaldi, L.; Zarek, P.E.; Horton, M.; Powell, J.D. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol. Immunother. 2011, 61, 917–926.
- Kjaergaard, J.; Hatfield, S.; Jones, G.; Ohta, A.; Sitkovsky, M. A2A Adenosine Receptor Gene Deletion or Synthetic A2A Antagonist Liberate Tumor-Reactive CD8+ T Cells from Tumor-Induced Immunosuppression. J. Immunol. 2018, 201, 782–791.
- Stagg, J.; Divisekera, U.; McLaughlin, N.; Sharkey, J.; Pommey, S.; Denoyer, D.; Dwyer, K.M.; Smyth, M.J. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1547–1552.
- Stagg, J.; Divisekera, U.; Duret, H.; Sparwasser, T.; Teng, M.W.; Darcy, P.K.; Smyth, M.J. CD73-Deficient Mice Have Increased Antitumor Immunity and Are Resistant to Experimental Metastasis. Cancer Res. 2011, 71, 2892–2900.
- Guo, S.; Han, F.; Zhu, W. CD39—A bright target for cancer immunotherapy. Biomed. Pharmacother. 2022, 151, 113066.
- Bastid, J.; Regairaz, A.; Bonnefoy, N.; Déjou, C.; Giustiniani, J.; Laheurte, C.; Cochaud, S.; Laprevotte, E.; Funck-Brentano, E.; Hemon, P.; et al. Inhibition of CD39 Enzymatic Function at the Surface of Tumor Cells Alleviates Their Immunosuppressive Activity. Cancer Immunol. Res. 2015, 3, 254–265.
- Yan, J.; Li, X.-Y.; Aguilera, A.R.; Xiao, C.; Jacoberger-Foissac, C.; Nowlan, B.; Robson, S.C.; Beers, C.; Moesta, A.K.; Geetha, N.; et al. Control of Metastases via Myeloid CD39 and NK Cell Effector Function. Cancer Immunol. Res. 2020, 8, 356–367.
- Li, J.; Chen, L.; Billedeau, R.J.; Stanton, T.F.; Chiang, J.T.P.; Lee, C.C.; Li, W.; Steggerda, S.; Emberley, E.; Gross, M.; et al. Discovery of a Series of Potent, Selective, and Orally Bioavailable Nucleoside Inhibitors of CD73 That Demonstrates In Vivo Antitumor Activity. J. Med. Chem. 2022, 66, 345–370.
- Seitz, L.; Jin, L.; Leleti, M.; Ashok, D.; Jeffrey, J.; Rieger, A.; Tiessen, R.G.; Arold, G.; Tan, J.B.L.; Powers, J.P.; et al. Safety, tolerability, and pharmacology of AB928, a novel dual adenosine receptor antagonist, in a randomized, phase 1 study in healthy volunteers. Investig. New Drugs 2018, 37, 711–721.
- Cecchini, M.; Krishnan, K.; Giafis, N.; Scott, J.; Quah, C.S.; Bendell, J.C. ARC-9: Phase Ib/II study to evaluate etrumadenant (AB928)-based treatment combinations in patients with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2021, 39, TPS150.
- Fong, L.; Hotson, A.; Powderly, J.D.; Sznol, M.; Heist, R.S.; Choueiri, T.K.; George, S.; Hughes, B.G.; Hellmann, M.D.; Shepard, D.R.; et al. Adenosine 2A Receptor Blockade as an Immunotherapy for Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020, 10, 40–53.
- Chiappori, A.A.; Creelan, B.; Tanvetyanon, T.; Gray, J.E.; Haura, E.B.; Thapa, R.; Barlow, M.L.; Chen, Z.; Chen, D.T.; Beg, A.A.; et al. Phase I Study of Taminadenant (PBF509/NIR178), an Adenosine 2A Receptor Antagonist, with or without Spartalizumab (PDR001), in Patients with Advanced Non–Small Cell Lung Cancer. Clin. Cancer Res. 2022, 28, 2313–2320.
- Lim, E.A.; Bendell, J.C.; Falchook, G.S.; Bauer, T.M.; Drake, C.G.; Choe, J.H.; George, D.J.; Karlix, J.L.; Ulahannan, S.; Sachsenmeier, K.F.; et al. Phase Ia/b, Open-Label, Multicenter Study of AZD4635 (an Adenosine A2A Receptor Antagonist) as Monotherapy or Combined with Durvalumab, in Patients with Solid Tumors. Clin. Cancer Res. 2022, 28, 4871–4884.
- Young, A.; Ngiow, S.F.; Barkauskas, D.S.; Sult, E.; Hay, C.; Blake, S.J.; Huang, Q.; Liu, J.; Takeda, K.; Teng, M.W.; et al. Co-inhibition of CD73 and A2AR Adenosine Signaling Improves Anti-tumor Immune Responses. Cancer Cell 2016, 30, 391–403.
- Luke, J.J.; Powderly, J.D.; Merchan, J.R.; Barve, M.A.; Hotson, A.N.; Mobasher, M.; Kwei, L.; Luciano, G.; Buggy, J.J.; Piccione, E.; et al. Immunobiology, preliminary safety, and efficacy of CPI-006, an anti-CD73 antibody with immune modulating activity, in a phase 1 trial in advanced cancers. J. Clin. Oncol. 2019, 37, 2505.
- Li, X.-Y.; Moesta, A.K.; Xiao, C.; Nakamura, K.; Casey, M.; Zhang, H.; Madore, J.; Lepletier, A.; Aguilera, A.R.; Sundarrajan, A.; et al. Targeting CD39 in Cancer Reveals an Extracellular ATP- and Inflammasome-Driven Tumor Immunity. Cancer Discov. 2019, 9, 1754–1773.
- Young, A.; Mittal, D.; Stagg, J.; Smyth, M.J. Targeting Cancer-Derived Adenosine:New Therapeutic Approaches. Cancer Discov. 2014, 4, 879–888.
- TTerp, M.G.; Olesen, K.A.; Arnspang, E.C.; Lund, R.R.; Lagerholm, B.C.; Ditzel, H.J.; Leth-Larsen, R. Anti-Human CD73 Monoclonal Antibody Inhibits Metastasis Formation in Human Breast Cancer by Inducing Clustering and Internalization of CD73 Expressed on the Surface of Cancer Cells. J. Immunol. 2013, 191, 4165–4173.