2. Genome Mining for Key Enzymes in Biosynthesis Gene Clusters
Genome mining is a bioinformatic approach used to discover and analyze the biosynthesis genetic potential of natural products in the genome of organisms, such as plants, bacteria, and fungi. The most common genome mining approach is to apply computational algorithms to analyze biosynthetic gene clusters (BGCs), targeting known enzymatic or gene functions, particularly core biosynthesis enzymes and specific structural unit biosynthesis enzymes. The underlining rationale is that natural products are assembled, tailored, and regulated by strikingly conservative BGCs. These BGCs are responsible for composing production lines in natural organisms such as microbes, fungi, and plants [
8].
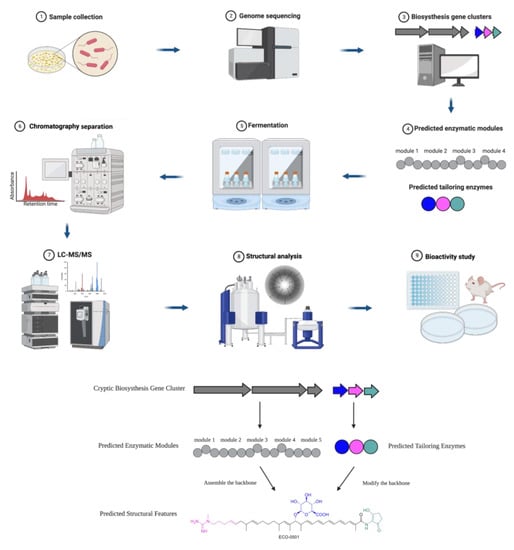
A typical roadmap of genome mining is elucidated in
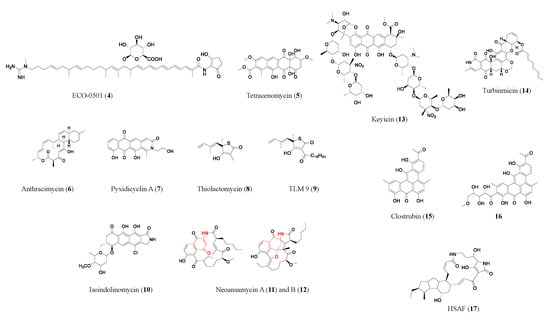
Scheme 1, which represents the discovery of a novel antibiotic through the excavation of bacterial genomes. The natural compound producers were collected from various sources and genomes were sequenced and analyzed for BGCs. Then, researchers used computational algorithms to predict the enzymatic modules and post-assembly tailoring enzymes, as exemplified by the discovery of the novel antibiotic ECO-0501 [
9]. The producer strain
Amycolatopsis orientalis ATCC 43491 was known to be a vancomycin-producing strain. A whole genome analysis indicated it harbors over ten BGCs besides vancomycin. Next, researchers expressed and characterized one of the clusters. A fermentation and chromatography-based separation yielded purified compound(s). Subsequently, structure elucidation through mass spectrometry (MS) and nuclear magnetic resonance spectroscopy (NMR) revealed the structure of a new glycosidic polyketide ECO-0501(
4). ECO-0501 exhibits strong antibacterial activity against MRSA (MIC: 4.8 μM) and vancomycin-resistant
Enterococci (VRE) (MIC: 10 μM) [
9].
Scheme 1. Roadmap of genome-mining-guided novel natural product discovery. The typical steps of genome mining for novel antibiotics are as follows. (1) Natural compound producers are collected from various sources. (2) The genomes of natural compound producers are sequenced. (3) The sequenced genomes are analyzed for biosynthesis gene clusters (BGCs). (4) Computational algorithms are used to predict enzymatic modules and post-assembly tailoring enzymes. (5) The producers of interest (such as bacteria and fungi) are often fermented to express the target compounds. (6) Chromatography-based separation is deployed to obtain purified compounds. (7) The target compounds are characterized through analytical methods such as HPLC and MS. (8) The chemical structures are elucidated by NMR or single crystal diffraction. (9) The biological activities of the compounds are investigated. These steps of excavating a bacterial genome for BGCs leading to the discovery of a novel antibiotic were exemplified by ECO-0501, a glycosidic polyketide.
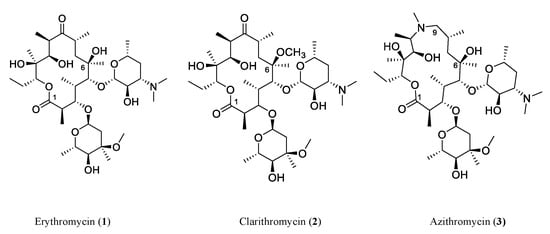
2.1. Polyketides
Polyketides are biosynthesized by polyketide synthases (PKSs) using acyl-CoA as a precursor. Generally, bacterial type I PKSs have multiple enzymatic modules, each module only adds one unit of acyl-CoA to elongate the polyketide chain, and subsequent catalysis often leads to the formation of the macrocycle (e.g., erythromycin (
1) in
Figure 2). In contrast, bacterial type II PKSs act iteratively, with one enzymatic module being used repeatedly to grow the polyketide chain. Subsequent steps often lead to the formation of a polycyclic aromatic skeleton (e.g., tetracenomycin (
5) in
Figure 2) [
10]. The understanding of the biosynthetic rationale behind polyketides has come a long way; using this extensive information, guidelines for predicting cryptic polyketide structures and activity from genome sequences have been developed [
11].
Figure 2. Representative antibiotics of the polyketide class. Red marks components derived from 3-amino-5-hydroxybenzoic acid (AHBA), the starter unit in neoansamycins (11, 12). HSAF (17) is a heat-stable antifungal factor, a drug candidate with exemplary bioactivities and a novel antifungal mode of action.
Besides the example of type I polyketide ECO-0501 researchers discussed above, additional compounds were discovered through a similar approach. Anthracimycin (
6), a promising type I polyketide antibiotic with a unique structure, produced by marine-derived
Streptomyces sp. CNH365, has shown potent activity against anthrax-causing Bacillus anthracis UM23C1–1 (MIC: 0.079 μM) and various antimicrobial-resistance pathogens, such as MRSA (MIC ≥ 0.63 μM) and vancomycin-resistant
S. aureus (MIC ≥ 0.63 μM) [
12]. Anthracimycin has been shown to protect animals from MRSA-induced mortality in a murine model of infection, and macromolecular synthesis studies suggest that it may inhibit RNA and/or DNA synthesis of bacteria [
13]. Similarly, the PKS gene cluster in the genome of the myxobacterium
Pyxidicoccus fallax was investigated, and genes encoding pentapeptide repeat proteins, known to confer resistance to topoisomerase inhibitors, were found in the cluster. Its activation in the natural host and heterologous expression allowed the discovery of pyxidicyclines (e.g., pyxidicyclin A (
7)) and precursor anthraquinones [
14].
The heterologous expression approach is often combined with the resistance gene screening approach. Using these two combined approaches, a PKS–NRPS hybrid gene cluster led to the discovery of a series of thiotetronic acid natural products, including the previously known fatty acid synthase inhibitor thiolactomycin (
8), which has moderate activity against MRSA,
Mycobacterium tuberculosis,
Plasmodium falciparum, and
Francisella tularensis [
15]. In a subsequent study, the researchers synthesized structural analogs of thiolactomycin which showed enhanced activity (e.g., TLM 9 (
9) showed a 75-fold improvement against MRSA) (Bommineni, 2016). A polyketide with a unique tetracyclic skeleton, isoindolinomycin (
10), has a BGC which is mutated in the wild-type
Streptomyces sp. SoC090715LN-16. Through screening of rifampicin-resistant mutants and activation of the biosynthetic gene cluster, isoindolinomycin was discovered and characterized [
16]. Tetracyclic-type antibiotics, which inhibit bacterial growth by specifically binding to bacterial ribosomal subunits, currently face efflux pump-type resistance due to overuse [
17]. Isoindolinomycin may offer new target binding sites and overcome the current antimicrobial resistance issue.
Other than examining the whole PKS clusters, another common methodology is to screen for key biosynthesize units. The synthase genes for 3-amino-5-hydroxybenzoic acid (AHBA), the starter unit in ansamycin-type antibiotics, were screened in a pool of actinobacteria [
18]. The genome of
Streptomyces sp. LZ35, an AHBA synthase-positive strain, was further analyzed for novel BGCs. Multiple attempts were then made to successfully activate the silent neoansamycin BGC through transcriptional regulation factors, leading to the discovery of neoansamycins (
11,
12) [
19,
20].
Sometimes silent BGCs can be activated through adjustment of the microbial culture conditions. Keyicin (
13), a poly-nitroglycosylated anthracycline antibiotic, was discovered by co-culturing two marine bacteria from the otherwise silent PKS gene cluster in the producing strain
Micromonospora sp. In addition to its possible interspecies crosstalk function, keyicin exhibits selective antimicrobial activity against Gram-positive bacteria including
Rhodococcus sp. and
Mycobacterium sp. and it does not cause nucleic acid damage [
21].
Moreover, genomics leveraging analytical techniques (e.g., LC-MS, HPLC, NMR, CryoEM, etc.) [
22,
23,
24,
25,
26] has been used to profile the metabolic phenotypes of microbes for prospecting antibiotics. A discovery platform was built that applies genomics, metabolomics, and antimicrobial activity screening of LC-MS-based metabolomic arrays from 174 marine animal isolated bacteria [
27].
Micromonospora sp. WMMC-415’s hidden chemical diversity was highlighted and prioritized by its potent activity against
Candida albicans and led to the discovery of turbinmicin (
14), a broad-spectrum antifungal drug candidate that targets multidrug-resistant fungi with a high safety profile and a highly selective mechanism of action [
27].
Genome mining of BGCs is often combined with activity-guided separation. Clostrubin (
15), a highly unsaturated aromatic polyketide from an obligate anaerobe
Clostridium beijerinckii, demonstrated pronounced activity against various pathogenic Gram-positive bacteria, including MRSA (MIC: 0.12 µM), VRE (MIC: 0.97 µM), and mycobacteria (MIC: 0.48 μM), and moderate antiproliferative and cytotoxic effects [
28]. In another study, clostrubin and its analog (
16) were found to be produced by
Clostridium puniceum functioning as antibiotics against other plant pathogens, giving the anaerobic bacteria an advantage to compete in an oxygen-rich plant environment [
29]. Clostrubins are an exemplary case of mining the genomic biosynthesis potential in combination with traditional culture condition screening and/or molecular biology tools, leading to the rapid discovery of polyketide antibiotics.
2.2. Nonribosomal Peptides
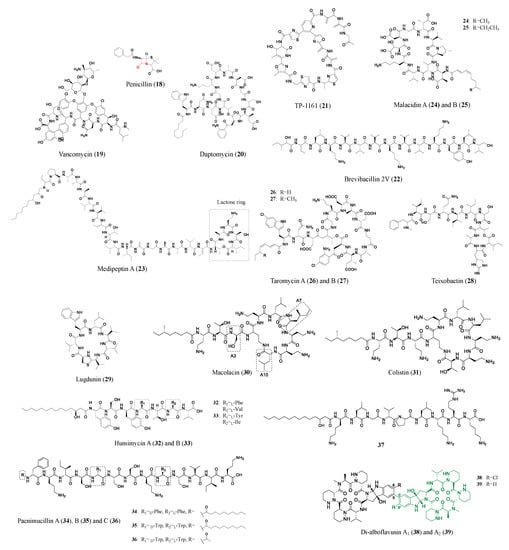
Distinguished from peptides and proteins synthesized from the ribosomal pathway, nonribosomal peptides are assembled by modular machinery consisting of large, selective multifunctional enzymes called nonribosomal peptide synthetases (NRPS). The products of these machineries are many successfully marketed drugs (e.g., penicillin (
18), vancomycin (
19), and daptomycin (
20) shown in
Figure 3) [
30]. Over the last few decades, the complicated production process of nonribosomal peptides has been the subject of biochemical and structural biology investigations, offering new compounds with pharmaceutical value.
Figure 3. Representative antibiotics of the nonribosomal peptide class. The β-lactam ring of penicillin (18) is marked in red. The amino acids at A3, A7, and A10 positions of the cyclic polypeptide of Colistin (31), also known as polymyxin E, are replaced to generate macolacin (30). Green colored structures are the monomers of di-alboflavusins (38, 39).
Straightforwardly, conserved genes of BGCs can be screened to mine new nonribosomal peptides with pharmaceutical value. Twenty-seven marine actinomycete isolates were subjects for NRPS and PKS gene screening, and coupled with a bioactivity assay, this led to the identification of
Nocardiopsis sp. TFS65-07. Further isolation and examination determined the active compound to be TP-1161 (
21), a new macrocyclic thiopeptide with three thiazole and two oxazole groups. TP-1161 displayed superior activity against a panel of Gram-positive pathogens including VRE (MIC: 0.84 μM), and the mode of action for thiopeptide is yet to be determined [
31]. Similarly, genome mining of NRPS gene clusters in
Brevibacillus laterosporus DSM 25 led to the discovery of Brevibacillin 2V (
22), which has a strong antimicrobial activity against difficult-to-treat antimicrobial-resistant Gram-positive pathogens with low hemolytic activities and cytotoxicities to eukaryotic cells. When combined with marketed antibiotics, Brevibacillin 2V demonstrates a synergistic effect against Gram-negative pathogens and shows a high stability in human plasma, most likely because of the inclusion of D-amino acids and non-canonical amino acids [
32]. Likewise, a cyclic lipopeptide medipeptin A (
23) was isolated from the microbiome of a tomato plant in the Netherlands. Researchers took a sample from the plant and isolated
Pseudomonas bacteria, the genomic DNA of which was then analyzed for biosynthetic gene clusters. Medipeptin A was isolated and showed good activity against multiple pathogens. Medipeptin A’s antibiotic capabilities come from its binding with lipoteichoic acids and by forming pores in the bacterial membrane [
33].
Many nonribosomal peptides encoded by unculturable bacteria genomes can be obtained in the heterologous expression hosts. Malacidins (
24,
25), which are calcium-dependent nonribosomal lipopeptides, target the cell wall biosynthesis of multidrug-resistant pathogens. The malacidin gene cluster was captured from desert soil metagenomics using primers targeting calcium-binding motifs. To avoid rediscovery, a phylogeny tree of the calcium-dependent antibiotic gene was generated to guide the selection of a novel malacidin gene cluster. Heterologous expression yielded new antibiotic malacidins, whose activity spectrum against multiple drug-resistant pathogens was evaluated and the mode of action was discussed [
34]. Similarly, taromycin A (
26) and B (
27) are nonribosomal lipopeptides whose BGCs highly resemble the daptomycin gene cluster. The 67 kb otherwise silent taromycin BGC was directly cloned and refactored into the model expression host
Streptomyces coelicolor. Taromycins show moderate activity against multiple daptomycin-resistant strains, demonstrating an alternative option for a new antibiotic [
35,
36]. Although the heterologous expression of NRPS clusters often proceeds with difficulty due to the giant gene cluster size (30–100 kb), the success of malacidins and taromycins demonstrated new avenues for antibiotic discovery.
Another breakthrough method to excavate NPs from unculturable bacteria was to grow them directly in their native environments, such as soil and seawater. Teixobactin (
28) was discovered using a device that allows source bacteria to grow between semipermeable membranes immersed in the original soil sample, enabling nutrients, and signaling molecules to promote growth. Teixobactin represents a new class of antibiotics with excellent activity against Gram-positive pathogens and a non-detectable resistance [
37]. In addition, new antibiotics may come from previously overlooked sources such as the animal microbiome. Lugdunin (
29) is a thiazolidine-containing nonribosomal peptide that is produced by the human microbiota
Staphylococcus lugdunensis. Ludunin displays a broad spectrum of activity against Gram-positive pathogens and prevents
S. aureus from colonizing. Researchers also observed that
S. lugdunensis nasal colonization was linked to a notably low
S. aureus carriage rate, indicating that lugdunin or lugdunin-producing commensal bacteria may prevent staphylococcal infections [
38].
NP bioinformation-guided and organically synthesized peptides are a new approach to rapidly obtaining antimicrobial agents. Macolacin (
30), an analog of the nonribosomal peptide antibiotic colistin (
31), is a synthetic bioinformatic natural product generated by chemical synthesis from the BGC-guided approach. Through an extensive search of bacteria genomes for colistin-like BGCs, a BGC from
Paenibacillus xylanexedens mac was projected to produce a structurally divergent colistin congener. Subsequential chemical syntheses produced macolacin and its derivatives, which exhibit superior activity over colistin against multidrug-resistant pathogens, including drug-resistant
Acinetobacter baumannii and colistin-resistant
Neisseria gonorrhoeae [
39]. A similar chemical synthesis approach was applied to produce humimycins, which are MRSA active antibiotics, from human-associated bacteria NRPS clusters. Two NRPS clusters from the genomes of
Rhodococcus equi and
Rhodococcus erythropolis predicted the amino acid sequences of humimycins A (
31) and B (
32), respectively [
40]. Using synthetic endeavors, more nonribosomal peptides (Paenimucillins (
34–
36)) and an antifungal peptide (
37) were discovered. Paenimucillin C (
36) inhibits multidrug-resistant
Acinetobacter baumannii clinical isolates and sterilizes multidrug-resistant
A. baumannii infection with no indication of a rebound in a murine model [
41,
42].
The synthetic bioinformatic NP approach bypasses complicated gene cluster activation in the original host, avoids establishing a heterologous expression system for large gene fragments, skips the time-consuming fermentation extraction and isolation, and alleviates the difficulties of complicated peptide structure elucidation. However, synthetic peptides usually cannot generate the final products of the BGCs, partly due to the accuracy of the computational prediction of versatile and complicated post-assembly tailoring enzymes. Despite the limitations, this approach offers adequate peptide skeletons for a structure–activity relationship exploration of the analogs. Among post-assembly tailoring enzymes, cytochrome P450s successfully generated activity-improved nonribosomal peptides. Inspired by the significant antibacterial property enhancement of the dimeric products, Guo et al. searched Streptomyces genomes for tailoring enzyme cytochrome P450s with the function of dimerizing structural similar nonribosomal peptides [
43]. The overexpression of two cytochrome P450s in
Streptomyces alboflavus sp. 313 led to the generation of dimeric nonribosomal peptide di-alboflavusins. Di-alboflavusin A
1 (
38) and A
2 (
39) demonstrate a 10–100-fold improvement in antibacterial activity against multiple MRSA strains compared with the counterpart monomers (marked green in
Figure 3) [
43].
Figure 3 shows the nonribosomal peptides discussed in this section.
2.3. Ribosomally Synthesized and Post-Translationally Modified Peptides
Ribosomally synthesized and post-translationally modified peptides (RiPPs) present high complexity and miscellaneous bioactivities for pharmacological applications [
44]. The intricate RiPP molecular structures arise from diverse peptide skeletons and complex post-assembly modifications, generating linear peptides, cyclic peptides, lanthipeptides, lasso peptides, thiopeptides, lipopeptides, glycopeptides, etc., presenting many unique drug candidates for antimicrobial-resistant infections [
45].
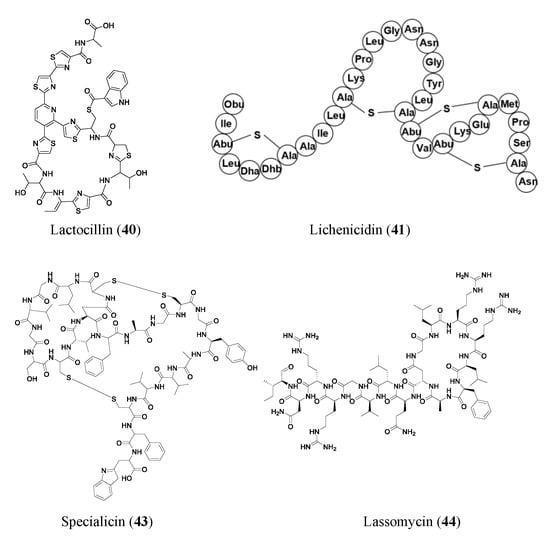
Many RiPPs are found through simple genome mining for BGCs. Lactocillin (
40), a thiopeptide antibiotic, is a classic example of a product of massive genome data mining of bacterial BGCs from unconventional biological systems. Lactocillin BGC from a vaginal microbiota genome was pinpointed out of 3118 BGCs from human-associated bacteria genomes. Lactocillin displayed potent activity against vaginal pathogens. Additionally, metatranscriptomic sequencing data show that lactocillin is expressed in the human vagina in vivo, suggesting vaginal microbiota may inhibit pathogenic bacteria and prevent vaginal infections through the expression of lactocillin-type thiopeptides [
46].
The novel two-peptide lantibiotic lichenicidin (
41) was isolated from Bacillus licheniformis D13 and was found using NCBI-BLAST to look through the B. licheniformis genome to detect gene clusters that could produce novel lantibiotics. Lantibiotics are a class of RiPP antibiotics characterized by post-translational addition of the thioether amino acids lanthionine and methyllanthionine [
47]. Lichenicidin has activities against Bacillus megaterium, Bacillus subtilis,
S. aureus, Micrococcus luteus, and Rhodococcus spp. [
48]. Another RiPP found through genome mining of BGCs is thiomuracin (
42). Thiomuracin is a thiopeptide found by genome mining of the bacterium
Thermobispora bispora. The peptide has strong activities against methicillin-resistant
Staphylococcus aureus USA300 (MIC of 0.18 μM) and vancomycin-resistant
Enterococcus faecium U503 (MIC of 0.046 μM). The peptide also has an MIC of 1.5 μM against spore-forming Bacillus anthracis str. Sterne. Thiomuracin is post-translationally modified into a cyclic structure that has inhibitory effects on bacterial protein synthesis [
49].
Other novel RiPPs were found through targeted genome mining, where an analog or type of gene cluster was searched for. Specialicin (
43) is a lasso peptide that was found by searching the Streptomyces specialis genome for analogs of known peptides. A BLAST search was run, looking for BGCs that were similar to those of siamycin. Specialicin was then found, isolated, and tested, which revealed an MIC of 3.75 μM against M. luteus [
50]. Another lasso peptide, Lassomycin (
44), showed promising activities against
Mycobacterium tuberculosis (0.41–1.65 μM), including many resistant strains (0.21–1.65 μM). Lassomycin was found in an extract of
Lentzia kentuckyensis. It is an interesting lasso peptide because its N-terminus tail does not string through its macrolactam ring similar to many other lasso peptides [
51].
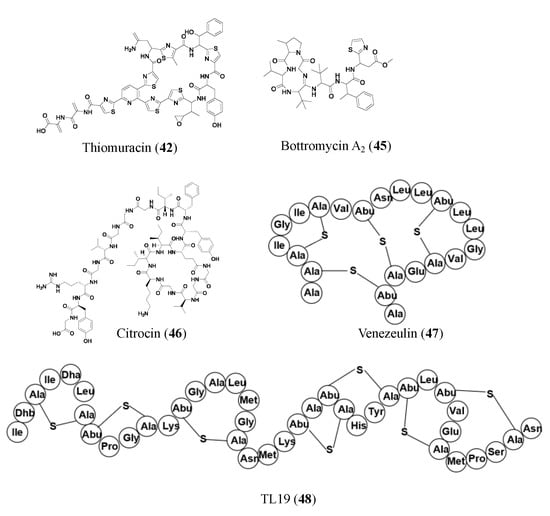
Promoter engineering and synthetic modular regulatory elements have successfully turned on silent BGCs for new NP production and constructed overproducing strains of known antibiotics [
52]. New bottromycin derivatives with 5–50-fold higher titers of bottromycin A
2 (
45) were generated by replacing native promoters with randomized constitutive synthetic promoters. The expression of the bottromycin gene cluster is decoupled from intricate native regulatory networks via promoter engineering and heterologous expression. Additionally, this tactic enhanced the production of bottromycins, a class of macrocyclic peptides with antimicrobial activities against multidrug-resistant bacteria [
53,
54].
Some RiPPs are heterologously expressed in a different host than the gene they are found in, one such compound is citrocin (
46). Citrocin is an antimicrobial lasso peptide found in both
Citrobacter pasteurii and
Citrobacter braakii. It was heterologously expressed in Escherichia coli and then isolated. Its structure was determined, and MIC values were calculated for its antimicrobial activities against a handful of species. Citrocin displayed an MIC ranging from 16–100 μM for
E. coli species with 16 μM for enterohemorrhagic
E. coli O157:H7 strain TUV93-0. The compound also displayed an MIC of 1000 μM against
Salmonella enterica serovar Newport and 125 μM against a clinical isolate of Citrobacter [
52]. An additional compound, venezeulin (
47), was found by genome mining for lantibiotic synthetases [
55]. Venezeulin, isolated from
Streptomyces venezuelae, is heterologously produced in
E. coli BL21 [
56].
Similar to synthetic bioinformatic nonribosomal peptides, RiPPs can be synthetically produced. TL19 (
48) is an antibiotic peptide (MIC of 0.9 to 15 μM against E. faecium) made by synthetically combining the N-terminus portion of nisin and the C-terminus portion of haloduracin α (HalA1 is one of the two peptides that make up the lanthipeptide haloduracin) [
56]. Nisin, an FDA-approved safe peptide food preservative with potential for clinical use, is a well-studied 34-residue lantibiotic that is produced by
Lactococcus lactis, where its synthesis is regulated by a two-component regulatory system. Haloduracin is a two-peptide antibiotic. Both HalA1 and Nisin contain a lipid II binding site [
57]. The aforementioned representative RiPP antibiotics are shown in
Figure 4.
Figure 4. Representative antibiotics of ribosomally synthesized and post-translationally modified peptides. Lanthipeptides are displayed in amino acid sequences instead of chemical structures to highlight the thioether cross-linkages.
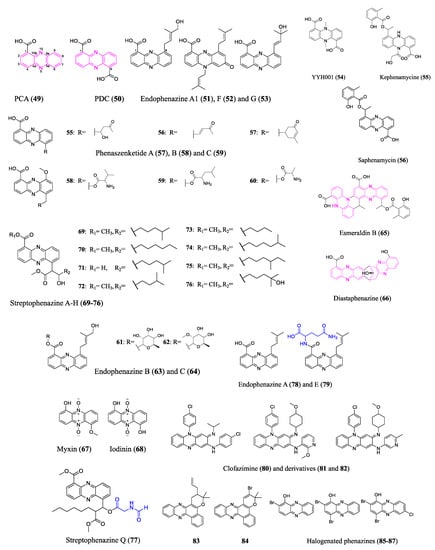
2.4. Phenazines
The fundamental structure of phenazines is composed of redox-active nitrogen tricyclic aromatic rings; however, they differ in the modification groups. Many phenazines have broad spectrum antimicrobial properties, making them prospective antibiotics [
58,
59,
60]. The biosynthesis of the phenazine core structure (marked pink in
Figure 5), which diverges from the shikimic acid pathway, has been well studied. Bacterial producers have a conserved set of phenazine biosynthesis genes (PHZ) that encode enzymes that convert chorismic acid into complex phenazine precursors phenazine-1-carboxylic acid (PCA) (
49) or phenazine-1,6-dicarboxylic acid (PDC) (
50) [
61]. Similar tactics have been employed to unearth new phenazines to those used for other classes of antibiotics discussed previously.
Figure 5. Representative compounds of the phenazine class. Pink marks the phenazine core structures in 49, 50, 65 and 66. Blue represents amino acid derived modification to 77 and 79. Phenazine-1-carboxylic acid (PCA, 49), also known as tubermycin B, has activity against M. tuberculosis. It is also a widely recognized natural fungicide in China that is effective and safe for the environment. Clofazimine (80) has been applied clinically to treat leprosy and multidrug-resistant M. tuberculosis. Endophenazine E (79) does not have an antibiotic effect, which could result from the heterologous host’s defense mechanism.
Phenazine BGC screening and bioactivity-guided isolation are two commonly combined approaches to excavate new phenazine antibiotics. A bioinformatics analysis of Kitasatospora sp. HKI 714 genome revealed phenazine BGC and the subsequent bioactivity-guided isolation revealed new terpenoid-substituted phenazines endophenazine A1 (
51), F (
52), and G (
53). Heine et al. analyzed the biological effects of the new endophenazines, mostly concentrating on their antibacterial properties against various mycobacteria and MRSA [
62]. A group applied the bioinformatic tool antiSMASH 6.0 [
63] to analyze the genome of
Amycolatopsis nigrescens DSM 44992, revealing one phenazine BGC as well as thirty other BGCs. An antimicrobial-activity-guided isolation revealed a novel phenazine compound YYH001 (
54), whose absolute structure was determined by crystallography. However, additional bioactivity assays did not find appreciable antibiotic properties of the purified YYH001. Researchers speculated that the superior activity against bacterial and fungal pathogens was brought by other BGC products of the crude extract. In fact, researchers successfully identified a family of novel polyketide antibiotics from a PKS gene cluster in
Amycolatopsis nigrescens. Besides the assessment of BGCs, researchers often use universal primers of conserved genes to find the microbes’ genetic potential for antibiotic production. This approach is still suitable for cost-effective and time-efficient screening of large numbers of microorganisms. Researchers used universal gene phzE primers to evaluate a pool of actinobacteria that a group isolated from unusual biosamples. The positive strain
Streptomyces sp. B23 isolated from a soil sample was selected for further investigation. A bioactivity-guided separation led to the discovery of a novel antibiotic kephenamycine (
55), along with several known phenazines (e.g., saphenamycin (
56)).
The diverse bioactivity of phenazines arises from a variety of modifications to the core. Shi et al. reported that the entomopathogenic bacterium
Xenorhabdus szentirmaii underwent multiple modifications of the phenazine core through two different BGCs, generating derivatives with phenazine–polyketide hybrid scaffolds (e.g., phenaszenketide A–C (
57–
59)) and phenazine–peptide hybrid scaffolds (e.g., pelagiomicin B (
60) and phenaszentine C (
61)). The two BGC pathways were connected via a common aldehyde intermediate altered by several enzymatic and non-enzymatic reactions. The evaluation of the antibiotic activity of the generated phenazine derivatives points to a highly efficient method for converting the phenazines with specialized activity against Gram-positive bacteria into broad spectrum antibiotics [
64]. However, the antibiotic activity of phenazines against pathogens was evaluated via diameters of inhibition zones in disc diffusion assays and lack of replications; MICs against antimicrobial-resistant pathogens need to be conducted to comprehensively assess their antibiotic properties. Other research discussed the antibacterial mode of action of a structurally similar phenazine–peptide
D-alanylgriseoluteic acid (
62), which induced the SOS response through the endogenous redox activity of the compound [
65]. Other types of phenazine antibiotic modification seen in nature include glycosylation (e.g., endophenazine B (
63) and C (
64)) [
66], dimerization (e.g., esmeraldin B (
65) and diastaphenazine (
66), two cores marked pink in
Figure 5) [
67,
68], and N-oxidation (e.g., myxin (
67) and iodinin (
68)) [
69], among others.
Activation of silent gene clusters poses one of the major challenges of genome mining for new antibiotics. Low doses of antibiotics may act as signaling molecules that can activate or modulate BGCs. Streptophenazines A–H (
69–
76), a family of new phenazines, were found in a marine Streptomyces isolate. The production pattern of streptophenazines depends on the different antibiotics added to the culture conditions which act as signaling molecules. Specifically, tetracycline induces streptophenazine F and G production and increases the yield of streptophenazine A–D, whereas bacitracin induces streptophenazine H production. The streptophenazines showed moderate activity against Bacillus subtilis as well as Staphylococcus lentus [
70]. Heterologous expression and promoter engineering may also activate silent phenazine BGCs. A remarkable chemical diversity of streptophenazines was produced by heterologous expression and refactoring promoters of a PHZ/PKS/NRPS hybrid gene cluster from
Streptomyces sp. CNB-09. Among the 112 isolated compounds, a streptophenazine analog containing an unprecedented N-formylglycine moiety (marked in blue in
Figure 5), streptophenazine Q (
77), demonstrated an effective antibiotic activity against MRSA and disease-causing group A
Streptococcus. However, the heterologous expression of phenazine BGCs can be challenging due to the antibiotic effect of the products on the heterologous host without a proper resistance mechanism [
71]. The endophenazine BGC in the original host (Streptomyces anulatus) produces a much higher yield of antibiotic endophenazine A (
78) than the heterologous host Streptomyces coelicolor M512. The authors found that the heterologous host strains accumulated a glutamine adduct (marked in blue in
Figure 5) endophenazine E (
79), which does not have antibiotic activity, and they speculated that glutamination could be a defense mechanism of the host [
72,
73].
Furthermore, NP-inspired organic synthesized phenazines exhibit extraordinary pharmaceutical value against antimicrobial-resistant pathogens and eradicate persistent pathogen biofilms. Clofazimine (
80) has been applied to treat multidrug-resistant Mycobacteria tuberculosis, may compete with an essential component in the mycobacterial electron transfer chain, and releases reactive oxygen species [
74]. Clofazimine derivatives (
81 and
82), which displayed a remarkable potency improvement in vitro and reduced lipophilic-associated side effects, were further examined for pharmacokinetic profiles in vivo [
75,
76,
77]. Coelho et al. examined the in vitro activity of several phenazine derivatives against rifampicin-resistant
M. tuberculosis and pan-susceptible
M. tuberculosis H37Rv and found that the allyl-pyran-group-containing phenazine (
83) showed an MIC of 2.2 μM and the bromo-pyran-group-containing phenazine (
84) showed an MIC of 16 μM [
78]. More excitingly, marine brominated phenazine (
85) inspired the synthesis of a series of halogenated phenazines (e.g.,
86–
87) whose structure–activity relationships were thoroughly investigated by Prof. Huigens and colleagues. Potent halogenated phenazine analogs successfully eradicated biofilms formed by persistent pathogens through rapid induction of iron starvation with minimal mammalian cytotoxicity and hemolysis. These findings informed and encouraged the design of rational prodrugs for pharmacological translation [
79,
80,
81,
82]. The structures of the aforementioned representative phenazines are shown in
Figure 5.
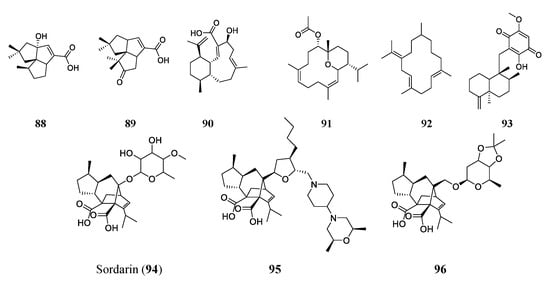
2.5. Terpenes/Terpenoids
Terpenes are often thought to be the largest class of natural products. They often come from plants but are also common in fungi and bacteria. Terpenes are made of repeating isoprene units and typically have carbons in multiples of five because of the five-carbon nature of isoprenes [
83]. Specifically, terpenoids are compounds made of isoprene units that may contain functional groups such as oxygen, while terpenes are hydrocarbons composed only of hydrogen and carbon atoms, but these terms are often used interchangeably in the literature [
84].
A BGC analysis can also indicate the genetic potential of producing terpene-type antimicrobials. Researchers sequenced the genome of actinobacteria
Streptomyces spp. NRRL S-4 and searched for pentalenolactone analogs using the antiSMASH platform. The results revealed six terpene BCGs. Two were determined to produce two sesquiterpenoids 1-deoxy-8α-hydroxypentalenic acid (
88) and 1-deoxy-9β-hydroxy-11-oxopentalenic acid (
89). These two compounds had MICs of 64 and 61 μM, respectively, against
S. aureus ATCC 25923 and 128 and 61 μM against
E. coli ATCC 25922, respectively [
85]. Another terpenoid found through genome mining is benditerpenoic acid (
90). The BGC of benditerpenoic acid, a novel bicyclic diterpenoid, was found in the genome of
Streptomyces sp. CL12-4. The compound was then heterologously expressed in
E. coli, isolated, and tested to find its MIC against
E. faecium (401 μM),
S. aureus (200 μM), MRSA (200 μM), multidrug-resistant
S. aureus (401 μM), and
B. subtilis (100 μM) [
86].
More often, terpenoids are found through the traditional approach of bioactivity screening of extracts or broths.
Sarcophyton trocheliophorum, a species of soft coral found in the Red Sea, has been found to produce five terpenoid compounds. Two of these compounds are novel and pyrane-based and were exemplified by sarcotrocheliol (
91). Sarcotrocheliol showed substantial antibiotic activity with MICs of 1.53 μM against
S. aureus and 3.06 μM against MRSA. Cembrene-C (
92) was also isolated from this coral and had impressive antifungal activities, with an MIC of 0.68 μM against
Candida albicans and
Aspergillus flavus [
87]. A marine sponge of the genus
Hippospongia found in Fiji produced the sesquiterpenoid quinone epi-ilimaquinone (
93). This natural product had antibiotic activities of 182 μM against MRSA, 91 μM against
S. aureus, 45 μM against VRE, and 364 μM against amphotericin-resistant
Candida albicans [
88].
Among many terpenes and terpenoids, one class of compounds has very distinct chemical properties. The diterpene sordarin (
94), a glycoside antibiotic with a tetracyclic diterpene core, was first discovered in the 1970s and isolated from
Sordaria araneosa. Sordarin has been studied increasingly since its discovery to allow for the discovery of more, similar antimicrobial compounds [
89,
90]. The antifungal agent targets elongation factor II and halts protein synthesis. Sordarin is the subject of many studies now, with the aim of finding new analogs through genome mining and synthesis. Kudo et al. studied the genome sequence of
Sodaria araneosa Cain ATCC 36386 to reveal the sordarin biosynthesis pathway. Understanding the biosynthesis of sordarin allows for the targeted synthesis of analogs. FR290581 (
95), a sordarin analog, was synthesized by modification of sordaricin. It has an enhanced activity (MIC of 0.781–1.56 μM) against
Candida species [
90]. Another analog, GR193663 (
96), was synthesized by fusing the sugar moiety, creating a new innovative structure that had increased antifungal activities. The new compound GR193663 showed superior activity (MICs at the nanomolar level) against
Candida species [
91]. The structures of the aforementioned terepenes/terpenoids are shown in
Figure 6.
Figure 6. Representative compounds of the terpene/terpenoid class. Sordarin (94) is a glycoside antifungal agent that targets elongation factor II and halts protein synthesis. Semi-synthetic analogs FR290581 (95) and GR193663 (96) with innovative structures have shown enhanced activities compared to the original sordarin.