Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
TDP-43 belongs to a family of heterogeneous nuclear ribonucleoproteins (hnRNP). Structurally, TDP-43 consists of a structured N-terminal domain (NTD) involved in physiological self-oligomerization, followed by two tandem RNA-recognition motifs (RRM), which bind to certain nuclear transcripts and therefore regulate important DNA/RNA metabolism functions.
- TDP-43
- biomarker
- neurodegeneration
- amyotrophic lateral sclerosis
1. Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are diagnosed based on clinical criteria. Often, this leads to a delay between the onset of symptoms and diagnosis up to one year in ALS and 5 years in FTD, narrowing the window where potentially disease-modifying therapies would likely be most effective [1][2][3][4][5]. In order to intervene early and effectively in those diseases, biomarkers reflective of the underlying core pathobiology are needed [6][7]. These biomarkers will serve to identify the right cohorts for therapeutic targets and to determine the efficacy of therapies and clinical trials more accurately [8].
Ubiquitinated cytoplasmic aggregates of the nuclear 43 kDa transactive-response, DNA-binding protein, TDP-43, is the core pathobiology in almost all cases (97%) of ALS [9]. TDP-43 pathology is also found in 50% of frontotemporal lobar degeneration (FTLD) cases, most commonly associated with the clinical syndrome of behavioral variant frontotemporal dementia (bvFTD), while the rest is associated with tau protein pathology [10][11][12]. The interest in TDP-43 proteinopathies has further broadened since TDP-43 neuropathology has been found in 30% of Alzheimer’s disease (AD) cases [13][14]. Neuropathological evidence of TDP-43 pathology in AD has recently been defined as limbic-predominant age-related TDP-43 encephalopathy (LATE), which can present itself with or without coexisting hippocampal sclerosis pathology [15].
The clinical heterogeneity ranging from pure motor (ALS) to cognitively impaired (ALS-FTD) or dementia (FTD, LATE) phenotypes that are associated with TDP-43 proteinopathy is striking and likely associated with a distinct topographical distribution of TDP-43 pathology in the brain [16][17]. Pathological TDP-43 in ALS predominantly involves the spinal cord and the motor cortex but can be more widespread in the frontal and temporal lobe structures [18]. In FTD, TDP-43 is mostly found in the orbitofrontal cortex, but can exceed into the frontal and temporal cortices before involving the motor system, visual cortex, and cerebellum [19]. In contrast, TDP-43 pathology in LATE is mostly localized to the hippocampus and amygdala, suggesting that a selective cellular and regional vulnerability contributes to the disease [15]. There is, indeed, increasing evidence that TDP-43 functionality can be shaped depending on cell type [20][21]. Different types of TDP-43 pathology can be found in ALS/FTD clinical phenotypes; for example, type B pathology with large cytoplasmic inclusions and few dendritic neurites is mostly associated with ALS and bvFTD [22]. The clinical heterogeneity among FTD (bvFTD or primary progressive aphasia (PPA)) and the clinical similarity of LATE to AD makes it difficult to accurately predict the underlying pathology in life.
2. TDP-43 Pathobiology Informed Biomarker Development
2.1. Cellular Homeostasis and Aggregation Propensity
TDP-43 belongs to a family of heterogeneous nuclear ribonucleoproteins (hnRNP) [23]. Structurally, TDP-43 consists of a structured N-terminal domain (NTD) involved in physiological self-oligomerization, followed by two tandem RNA-recognition motifs (RRM), which bind to certain nuclear transcripts and therefore regulate important DNA/RNA metabolism functions (Figure 1a) [24][25][26][27][28]. The RRMs are flanked by N- and C-terminal regions. The C-terminal end of TDP-43 is a conserved, glycine-rich region important for the function of alternative splicing, protein–protein interactions and to promote the formation of phase-separated RNA granules [29][30][31][32][33]. The C-terminal domain of TDP-43 is also affected by most ALS-associated genetic variants [34][35], which, together with changes in RNA levels, can enhance the formation of dynamic liquid-like condensates [36][37]. In addition, an intrinsically disordered (IDR) prion-like region within the C-terminus harbors a tendency to self-assemble (“sticky ends”) and to form different oligomeric states [38][39]. There is, therefore, a critical equilibrium between TDP-43′s physiological function and its soluble form [24]. TDP-43 preferentially binds to long UG-rich sequences, including its own mRNA, via N-terminal homo-dimerization and its C-terminal conserved region [27][40][41][42]. Many of these regions have been found to be selectively modified by a variety of post-translational modifications, which include phosphorylation, acetylation, and SUMOylation [43]. Many of these events are now known to act as drivers of the pathology mostly by modifying the phase separation properties of this protein [44][45].
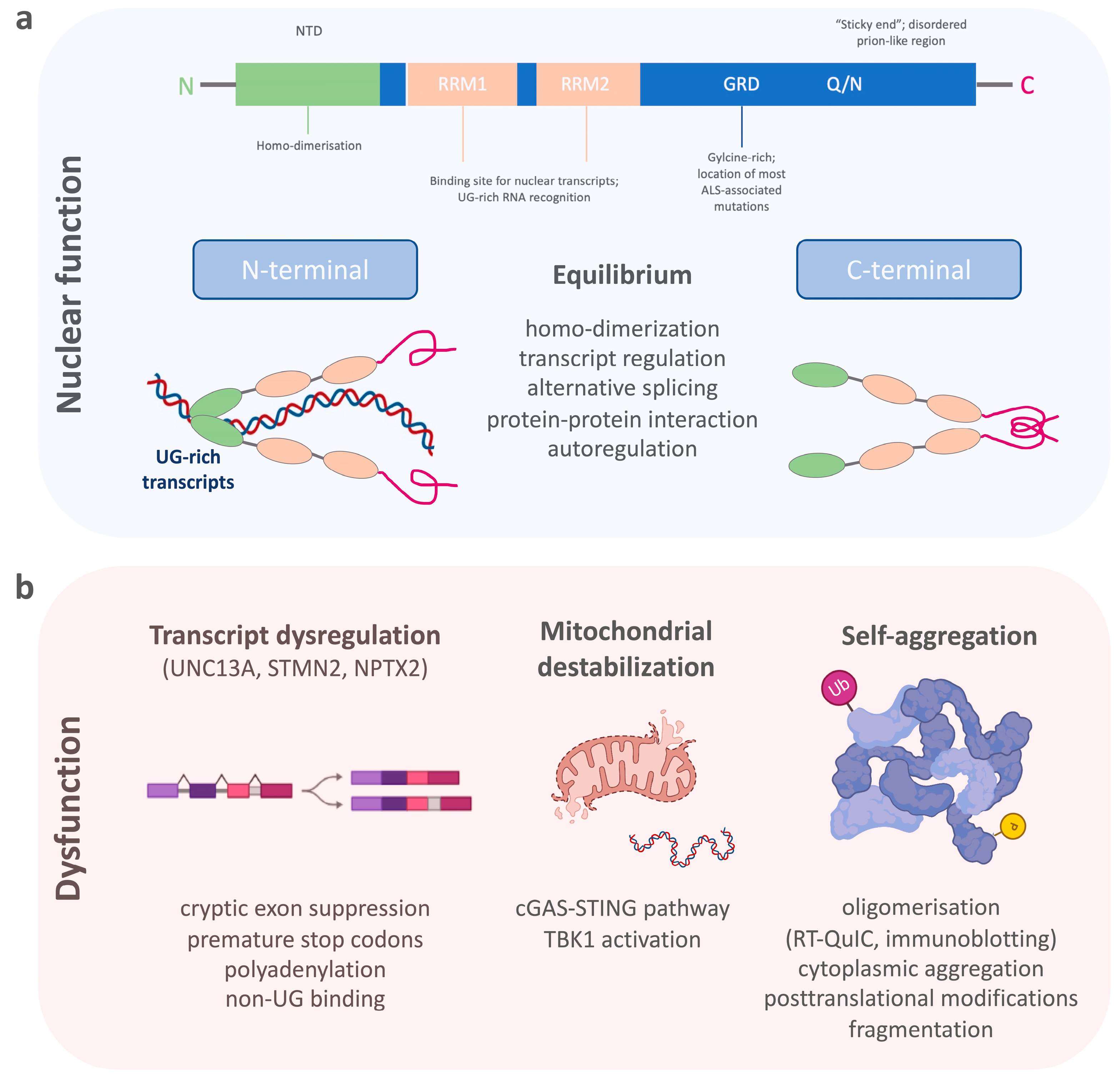
Figure 1. The schematic shows (a) the structure of TDP-43 and its importance for the physiological functions within the cell nucleus, and (b) the dysfunctional state of TDP-43 with the dysregulation of its nuclear function, destabilization of mitochondria, and its aggregation propensity.
Pathological processes that disturb the selective binding mechanism of TDP-43 enhance homonymous binding and self-aggregation, disturb its autoregulation, and lead to increased concentrations of TDP-43-aggravating protein accumulation (Figure 1b) [46][47]. Its potential for dimerization and oligomerization and the transcellular transmission of such TDP-43 oligomers has been shown in in vitro experiments [48][49][50]. Such self-aggregation and seeding propensities of misfolded proteins have been exploited for diagnostic purposes in prion disease, and later, a-synuclein and ß-amyloid pathologies using real-time quaking-induced conversion (RT-QuIC) [51][52][53][54]. Therefore, RT-QuIC for inducing TDP-43′s intrinsic aggregation propensity into non-amyloid, fibrillar aggregates, seemed a plausible technique for the detection of pathological TDP-43 species in patient biosamples [55][56][57]. A major requisite for developing RT-QuIC was achieved by producing functionally intact recombinant full-length and C-terminal fragmented TDP-43 species, which after mechanical and heat activation were able to seed amorphous TDP-43 aggregates or fibrillary structures in a measurable time frame [58]. This technique was then employed for cerebrospinal fluid (CSF) samples from patients with genetically confirmed TDP-43 proteinopathy and normal controls using the recombinant C-terminal fragment. A positive RT-QuIC reaction was obtained from TDP-43 positive samples with a high sensitivity of 94% and specificity of 85%. However, the limitations of this research were the false positive results, possibly originating from the presence of low levels of TDP-43 aggregates in healthy subjects, or by the heterogeneous presence of contaminants in the CSF, which needs to be tested in future studies. Another technique that uses aggregate formations of TDP-43 as a readout for TDP-43 proteinopathy is immuno-infrared sensor technology [59]. Here, TDP-43 is captured from CSF using oligoclonal antibodies targeting the aminoacid epitopes 130–275 of TDP-43. Bound TDP-43 is then measured by an infrared beam to assess the absorbance maximum indicative of the presence of an alpha-helical or β-sheet structure. The comparison of CSF from patients with ALS and Parkinson’s disease (PD), as well as neurological controls, demonstrated an increase in the β-sheet structures in CSF samples of ALS compared to PD and control samples. This approach displayed a diagnostic accuracy to discriminate ALS from PD (or controls) with 89% (89%) sensitivity and 77% (83%) specificity, with the caveat that this research lacked direct evidence of antibody-bound TDP-43 by immunoblotting. In summary, the self-aggregation propensity of TDP-43 has become a valuable tool for developing in vivo diagnostics; whether these techniques have the potential to be standardized for routine measurements requires further research.
2.2. Post Mortem TDP-43 Proteinopathy
The pathology of TDP-43 in ALS and FTD is characterized by its mislocalization from the nucleus to the cytoplasm of neurons and glia cells, where it is found post-translationally modified by phosphorylation and ubiquitination, as well as N-terminally cleaved into smaller C-terminal fragments (CTF) [9]. Neuropathological studies in ALS and FTD have shown that the phosphorylated form of TDP-43 is distributed in a relatively stereotyped spectrum of anatomical patterns, which has led to the definition of neuropathological stages [18][19]. In addition, the subtypes of characteristic TDP-43 staining were classified as different types (A–E) of TDP-43 proteinopathy, often characteristic for certain clinical syndromes within the ALS-FTD spectrum [60][61]. Both the regional localization of TDP-43 and the type of TDP-43 pathology are associated with a variable degree of cellular degeneration reaching from moderate whole-brain atrophy in ALS [62] to more severe focal frontal/temporal tissue loss in FTD [63].
Ultimately, a major breakthrough in neuropathology was achieved when the serial fractionation of post mortem brain tissue led to the extraction of insoluble proteins, which specifically recovered pathological TDP-43 from the affected brain regions [9]. A characteristic biochemical pattern of TDP-43 from the insoluble protein fraction includes a high molecular weight smear, which likely reflects phosphorylated and ubiquitinated TDP-43, phosphorylated full-length TDP-43 with a molecular mass size of 45–50 kDa and 60 kDa, and truncated forms at 24–26 kDa, identified as C-terminal fragments of TDP-43. C-terminal fragments and post-translational modifications of TDP-43 have been a prominent hypothesis proposed for TDP-43-associated neuronal toxicity and pathobiology [64][65]. Several studies therefore exploited the enrichment of pathological TDP-43 species from post mortem tissue in order to find a pathology-specific TDP-43 biomarker [9][10][66]. By mass spectrometry analysis, TDP-43 post-translational modifications or pathological peptide sequences were identified without the limitations of antibody-based methods. This approach enabled the identification of the pathological phosphorylation sites of TDP-43 predominantly located at its C-terminal glycine-rich domain [66][67]. These findings confirmed the disease-specificity of phosphorylated TDP-43 from previous studies using phosphorylation dependent TDP-43 antibodies [68]. Regarding phosphorylation, a recent important development has also occurred following the isolation of TDP-43 fibrils from post mortem brain samples and their analysis using mass-spec. This approach has allowed to start investigating possible PTM differences between the different types of TDP-43 pathology in GRN vs. C9ORF72 cases [69]. Further, a proteomic approach using post mortem tissue from different subtypes of TDP-43 proteinopathies allowed the identification of subtype-specific insoluble proteomes [70], while a similar approach using ALS post mortem tissue enabled the characterization of peptide sequences that were representative of TDP-43 protein fragments [66]. This work showed that peptides with a non-enzymatic N-terminal truncation site are representative of an endogenously cleaved C-terminal fragment, while less common peptides with a C-terminal truncation site represented N-terminal TDP-43 fragments. This was then taken forward to develop methods by targeted mass spectrometry for the absolute quantification of such fragment peptides [71][72][73]. Heavy isotope-labelled peptides of the reference peptides were used to absolutely quantify endogenous TDP-43 peptide levels. Using this method, it was shown that in the motor cortex of ALS patients with TDP-43 proteinopathy, the C:N-terminal peptide ratio of TDP-43 was significantly increased compared to both normal and other neurodegenerative disease control groups, suggesting a measurement for the enrichment of pathological C-terminal TDP-43 fragments in ALS [71]. The pathological cleavage of TDP-43 was further confirmed by measuring the N-terminal truncation site-specific TDP-43 peptides. This included the identification of a novel N-terminal truncation of a C-terminal fragment. Of note, the cleavage of TDP-43 was found in ALS, but also in the motor cortices of AD, that were later confirmed with LATE neuropathological changes [71]. In another work, multiple peptides spanning TDP-43 on structural and pathological relevant sites, including the N-terminus, both RNA-binding domains (residues 101–262) and the domains of TDP 35 kDa, and TDP 25 kDa fragments (residues 90–414 and 220–414) were developed [72]. Together with an enrichment process of functional RNA-binding TDP-43 using aptamers prior to mass spectrometric analysis, the sensitivity of detection was increased, and this enabled the improved quantification of the cleavage patterns of TDP-43 [73]. In conclusion, shifting the detection of TDP-43 on a proteotypic peptide level enables to form the basis for the absolute quantification of N-terminal and C-terminal TDP-43 fragments in future proteomic studies of complex matrices, such as blood and CSF, as well as the development of novel antibodies recognizing the pathological forms of TDP-43.
2.3. TDP-43 Loss of Function Mechanisms
While cytoplasmic aggregation with a toxic gain of function is a proposed mechanism of TDP-43 pathobiology, a second mechanism of toxicity is the nuclear loss of TDP-43 leading to altered RNA/DNA metabolism [74][75]. It is also suggested that the nuclear loss of TDP-43 could be an early event in the development of the disease preceding cytoplasmic TDP-43 aggregate formation [76][77]. TDP-43 acts as a splicing repressor of non-conserved intronic sequences, which, when disturbed, results in the inclusion of normally cryptic exons into mature RNAs (Figure 1b) [35]. This can lead to premature stop codons, premature polyadenylation, or the degradation of certain transcripts [78]. In line with this loss of function mechanism, a major alteration of the RNA levels of stathmin 2 (STMN2) and unc-13 homolog A (UNC13A) has been recently described [79][80]. Truncated and non-functional STMN2 mRNA, caused by premature polyadenylation resulting in an eightfold reduction of normal STMN2 levels, was found in the TDP-43 proteinopathy positive postmortem brain regions of FTD patients [81]. STMN2 is important for the stabilization of long axons in neurons and the loss of STMN2 has been associated with toxicity in ALS [82][83]. A similar pathology has been described for UNC13A, where TDP-43 depletion in neuronal cell lines lead to reduction of UNC13A transcripts through nonsense-mediated decay [80]. The repression of cryptic exon inclusion in UNC13A was present in neuronal nuclei with TDP-43 proteinopathy in post mortem brains from patients with ALS and FTD and the reduction of UNC13A protein after TDP-43 depletion was shown [84]. UNC13A is a synaptic protein important for glutamatergic neuronal transmission [85][86][87]. Pathological UNC13A polymorphisms in the same intronic region affected by mis-splicing have been associated with an increased risk in developing ALS/FTD, as well as a shorter survival [88][89][90][91]. In neurons overexpressing TDP-43 and post mortem brains with TDP-43 proteinopathy, the loss of nuclear TDP-43 was specifically associated with the upregulation of the synaptic neuronal pentraxin 2 (NPTX2) mRNA [92]. TDP-43 regulates the levels of NPTX2 mRNA through binding at its GU-rich region [93]. This function was shown to be disrupted in FTLD brain tissue where NPTX2 was also found aberrantly accumulated in neurons [92]. From a biomarker point of view, it is important to note that NPTX2 has also been found decreased in CSF samples of symptomatic FTD mutation carriers [94]. While most patients had TDP-43-associated chromosome 9 open reading frame 72 (C9ORF72) gene and granulin (GRN) gene mutations, NPTX2 was also lower in microtubule-associated protein Tau (MAPT) gene mutation carriers. Therefore, the specificity of NPTX2 as a biomarker for TDP-43 pathology is unclear and requires further investigation.
A further example of using a transcriptome-driven approach to develop non-invasive prognostic biomarkers for disease has been demonstrated for ALS [95]. In this approach, gene expression was derived from laser captured spinal motor neurons with TDP-43 pathology, and the data was bioinformatically processed into the modules associated with disease progression and upstream genetics. Measuring TDP-43 proteinopathy-dependent transcript alterations could therefore be a promising biomarker for discerning TDP-43 proteinopathies from other pathologies in life. In addition to the quantification of full-length protein levels, peptidomics is a promising tool to characterize protein isoforms and truncation signals that may arise from transcript alterations by including the search for unspecifically cleaved peptides. Data from proteomic studies of CSF samples so far have not been successful to detect STMN2 in either ALS patients or non-neurodegenerative diseases [96][97]. Alternative detection methods, such as hypothesis-driven targeted proteomics may therefore be considered in the future to enhance the sensitivity to detect a protein of interest by absolute quantification even in more complex matrices such as CSF [98].
2.4. TDP-43 Proteinopathy Induced Mitochondrial Dysfunction and Inflammatory Response
It is known that TDP-43 specifically interacts with mitochondrial proteins and alters mitochondrial dynamics [99]. Pathological TDP-43 fragments have also been found in mitochondrial protein fractions. However, these changes might be decreasing once neurodegeneration is severe, as recently shown in atrophic FTLD brain regions [100]. Nonetheless, many alterations in axonal mitochondrial transport and respiration have also been demonstrated in genetic models of ALS [101][102]. Most interestingly, in a cellular model of ALS cytoplasmic mislocalization of TDP-43 itself triggers its localization to mitochondria and leads to the release of mitochondrial DNA (Figure 1b) [103][104]. This process destabilizes the mitochondria by mitochondrial membrane permeabilization which eventually activates an inflammatory response via the cGAS/STING pathway. Other than the activation of the inflammatory cGAS/STING pathway, this leads directly to the activation of the TANK-binding kinase 1 (TBK1). In support, the TBK1 loss-of-function mutations have been linked to the development of both ALS and FTD TDP-43 proteinopathies [105]. While cGAS/STING activates TBK1, which then triggers a type 1 interferon response, mutations lead to haploinsufficiency of TBK1, meaning decreased TBK1 activity. Another interesting observation is that even loss of function of TBK1 seems to have an opposing effect in an ALS animal model [106]. The fact that TBK1 is directly associated with TDP-43 proteinopathy through the cGAS/STING pathway and can clinically lead to both diseases, ALS and FTD, and is eventually differentially regulated in early and late-stage disease, makes it an interesting pathway to follow for biomarker derivation. Inflammatory response, as well as evidence for inflammatory components that shape specific clinical and neuropathological patterns in the ALS-FTD-spectrum syndromes underpin the role of non-cell autonomous in ALS-FTD TDP-43 proteinopathy [107]. Among different inflammatory markers, chitinases, including chitotriosidase (CHIT1), chitinase-3-like-1 (CHI3L1), and chitinase-3-like-2 (CHI3L2), have been found to be significantly elevated in CSF of ALS patients compared to other neurodegenerative diseases and CHIT1-positive immunostaining was found in ALS spinal cord tissue, but not in the cortical tissue of ALS, FTD, or AD [108][109][110]. CHIT1 has also been found higher in the CSF of FTD patients with TDP-43 pathology [111]. However, the elevation of CSF CHIT1 levels in FTD are restricted to the clinical phase [112]. In addition, CHI3L1 in FTD correlates with frontal cognitive dysfunction [113][114].
Taken together the differential expression patterns of the chitinases in the ALS-FTD spectrum suggests that other factors, such as the severity of the disease, progression, or neurodegeneration pattern may account for differences rather than simply reflecting a reliable signal for TDP-43 proteinopathy.
2.5. Molecular Aspects of TDP-43 Pathology
TDP-43 pathology has been identified in a wide range of neurodegenerative diseases, both in cases inherited in a Mendelian pattern or sporadic patients, providing an exciting link between the pathophysiology of apparently sporadic and familial diseases. Generally, ~97% of all ALS and ~45% of FTLD cases involve TDP-43 aggregation [17][115]. In ALS, most sporadic patients and most patients carrying genetic variants, such as C9ORF72-, TARDBP-, OPTN-, UBQLN2-, and TBK1-ALS, show TDP-43 pathology [116][117]. Interestingly, in superoxide dismutase 1 (SOD1)-ALS, neuronal inclusions show immunopositivity for ubiquitin, but usually negativity for TDP-43. Similarly, in ALS due to genetic variants in the FUS RNA binding protein (FUS), a gene that is also encoding an RNA-processing protein involved in transcriptional regulation, TDP-43 positive inclusions are absent [118][119]. In FTD, TDP-43 pathology has been identified in Tau-negative sporadic cases (50%) as well as patients with genetic variants in, e.g., C9ORF72, GRN, and valosin-containing protein (VCP), but not in MAPT-FTD [17][116][117].
The discovery of key proteins might lead to the identification of candidate ALS-FTD genes, a prominent example being the identification of missense variants in TARDBP. Mutations in the gene encoding TDP-43 have, however, only being associated in less than 5% of ALS patients [34]. This is in line with the hypothesis that TDP-43 pathology may arise through multiple different mechanisms, emphasizing that ALS as a multistep disease, with fewer steps required in familial ALS [25][120]. On the other hand, the identification of mutated genes has a relevant impact on the development of novel disease biomarkers. For example, a loss of function mutation in cyclophilin A has been found in ALS and knocking out cyclophilin A in a mouse model led to the development of TDP-43 proteinopathy [121]. Cyclophilin A has also been found differentially expressed in ALS peripheral blood monocytes and plasma-derived extracellular vesicles [122][123]. These biomarkers could also be directly linked to the genetic defect, e.g., poly-GP in C9ORF72 expansion carriers or SOD1 protein concentrations in CSF of SOD1-ALS [124][125][126].
As alterations in RNA metabolism have emerged as one critical driver of ALS/FTD pathogenesis, the role of microRNA, small non-coding RNAs and key determinants of mRNA stability, has recently been studied [127][128]. Besides their important regulatory role in a variety of biological processes, miRNAs can also be released into the circulation by altered tissues as “fingerprints”, reflecting an activation of specific pathogenic pathways. Furthermore, they are easily detected in body fluids such as blood, CSF, or saliva, where they display remarkable stability. Interestingly, specific miRNA profiles have been identified in familial ALS, but also in pre-symptomatic mutation carriers [129]. At the level of a functional connection, it is known that the C-terminus of TDP-43 forms a complex with nuclear Drosha and cytoplasmic Dicer, which regulate miRNAs [130]. MiRNAs are gene expression modulators and influence cytoplasmic mRNA translation, indirectly giving evidence for a role of TDP-43 on cytoplasmic translation. Interestingly, TDP-43 has a direct effect on specific pri-miRNA processing and leads to differential expression of selected miRNAs [131][132], but the importance of these changes on disease onset and progression still remains to be largely evaluated.
In the future, however, additional types of non-coding RNAs could also be explored as ALS biomarkers. These include circRNAs that originate from back-splicing events during precursor mRNA processing and may act as miRNA sponges or to sequester RBP proteins, thus affecting protein and gene expression. Their usefulness as a biomarker mostly resides in the fact that circRNAs are very resistant to RNA exonucleases and are thus highly stable in cells. For example, a recent promising study of circRNAs in blood leukocytes of sporadic ALS patients has allowed to identify four circRNAs whose expression may be differentially affected in patients compared to controls [133].
Finally, long noncoding RNAs (lncRNAs) could also represent another possible type of RNA that could be easily detected in blood and CSF of patients. As the name suggests, lncRNAs are long RNAs that are not normally translated but can affect gene expression by affecting epigenetic and transcriptional profiles of their target genes. To this date, lncRNAs have not received a very high level of attention in ALS, but differential expression of several lncRNAs has been reported to occur in peripheral blood monocytes of ALS patients [134]. It should also be noted that TDP-43 has been described to also affect the processing of several of these molecules, such as NEAT1 and MALAT, and to affect the severity of the disease in animal models [135][136].
This entry is adapted from the peer-reviewed paper 10.3390/cells12040597
References
- Bott, N.T.; Radke, A.; Stephens, M.L.; Kramer, J.H. Frontotemporal dementia: Diagnosis, deficits and management. Neurodegener Dis. Manag. 2014, 4, 439–454.
- Cellura, E.; Spataro, R.; Taiello, A.C.; La Bella, V. Factors affecting the diagnostic delay in amyotrophic lateral sclerosis. Clin. Neurol. Neurosurg 2012, 114, 550–554.
- Cipriani, G.; Danti, S.; Nuti, A.; Di Fiorino, M.; Cammisuli, D.M. Is that schizophrenia or frontotemporal dementia? Supporting clinicians in making the right diagnosis. Acta Neurol. Belg. 2020, 120, 799–804.
- Draper, B.; Cations, M.; White, F.; Trollor, J.; Loy, C.; Brodaty, H.; Sachdev, P.; Gonski, P.; Demirkol, A.; Cumming, R.G.; et al. Time to diagnosis in young-onset dementia and its determinants: The INSPIRED study. Int J. Geriatr. Psychiatry 2016, 31, 1217–1224.
- Richards, D.; Morren, J.A.; Pioro, E.P. Time to diagnosis and factors affecting diagnostic delay in amyotrophic lateral sclerosis. J. Neurol. Sci. 2020, 417, 117054.
- Benatar, M.; Wuu, J.; McHutchison, C.; Postuma, R.B.; Boeve, B.F.; Petersen, R.; Ross, C.A.; Rosen, H.; Arias, J.J.; Fradette, S.; et al. Preventing amyotrophic lateral sclerosis: Insights from pre-symptomatic neurodegenerative diseases. Brain 2022, 145, 27–44.
- Kiernan, M.C.; Vucic, S.; Talbot, K.; McDermott, C.J.; Hardiman, O.; Shefner, J.M.; Al-Chalabi, A.; Huynh, W.; Cudkowicz, M.; Talman, P.; et al. Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2021, 17, 104–118.
- Turner, M.R.; Kiernan, M.C.; Leigh, P.N.; Talbot, K. Biomarkers in amyotrophic lateral sclerosis. Lancet Neurol. 2009, 8, 94–109.
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133.
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611.
- Mackenzie, I.R.; Neumann, M. Molecular neuropathology of frontotemporal dementia: Insights into disease mechanisms from postmortem studies. J. Neurochem. 2016, 138 (Suppl. 1), 54–70.
- Mackenzie, I.R.; Neumann, M.; Bigio, E.H.; Cairns, N.J.; Alafuzoff, I.; Kril, J.; Kovacs, G.G.; Ghetti, B.; Halliday, G.; Holm, I.E.; et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: Consensus recommendations. Acta Neuropathol. 2009, 117, 15–18.
- Amador-Ortiz, C.; Lin, W.L.; Ahmed, Z.; Personett, D.; Davies, P.; Duara, R.; Graff-Radford, N.R.; Hutton, M.L.; Dickson, D.W. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol. 2007, 61, 435–445.
- Josephs, K.A.; Whitwell, J.L.; Knopman, D.S.; Hu, W.T.; Stroh, D.A.; Baker, M.; Rademakers, R.; Boeve, B.F.; Parisi, J.E.; Smith, G.E.; et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 2008, 70, 1850–1857.
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019.
- Kawakami, I.; Arai, T.; Hasegawa, M. The basis of clinicopathological heterogeneity in TDP-43 proteinopathy. Acta Neuropathol. 2019, 138, 751–770.
- Tan, R.H.; Ke, Y.D.; Ittner, L.M.; Halliday, G.M. ALS/FTLD: Experimental models and reality. Acta Neuropathol. 2017, 133, 177–196.
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.; Van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 20–38.
- Brettschneider, J.; Del Tredici, K.; Irwin, D.J.; Grossman, M.; Robinson, J.L.; Toledo, J.B.; Fang, L.; Van Deerlin, V.M.; Ludolph, A.C.; Lee, V.M.; et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. 2014, 127, 423–439.
- Schweingruber, C.; Hedlund, E. The Cell Autonomous and Non-Cell Autonomous Aspects of Neuronal Vulnerability and Resilience in Amyotrophic Lateral Sclerosis. Biology 2022, 11.
- Susnjar, U.; Skrabar, N.; Brown, A.L.; Abbassi, Y.; Phatnani, H.; Consortium, N.A.; Cortese, A.; Cereda, C.; Bugiardini, E.; Cardani, R.; et al. Cell environment shapes TDP-43 function with implications in neuronal and muscle disease. Commun. Biol. 2022, 5, 314.
- Mackenzie, I.R.; Neumann, M.; Baborie, A.; Sampathu, D.M.; Du Plessis, D.; Jaros, E.; Perry, R.H.; Trojanowski, J.Q.; Mann, D.M.; Lee, V.M. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011, 122, 111–113.
- Krecic, A.M.; Swanson, M.S. hnRNP complexes: Composition, structure, and function. Curr. Opin. Cell Biol. 1999, 11, 363–371.
- Buratti, E.; Dork, T.; Zuccato, E.; Pagani, F.; Romano, M.; Baralle, F.E. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001, 20, 1774–1784.
- Al-Chalabi, A.; Calvo, A.; Chio, A.; Colville, S.; Ellis, C.M.; Hardiman, O.; Heverin, M.; Howard, R.S.; Huisman, M.H.B.; Keren, N.; et al. Analysis of amyotrophic lateral sclerosis as a multistep process: A population-based modelling study. Lancet Neurol. 2014, 13, 1108–1113.
- Lukavsky, P.J.; Daujotyte, D.; Tollervey, J.R.; Ule, J.; Stuani, C.; Buratti, E.; Baralle, F.E.; Damberger, F.F.; Allain, F.H. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct Mol. Biol. 2013, 20, 1443–1449.
- Afroz, T.; Hock, E.M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Pluckthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45.
- Wang, A.; Conicella, A.E.; Schmidt, H.B.; Martin, E.W.; Rhoads, S.N.; Reeb, A.N.; Nourse, A.; Ramirez Montero, D.; Ryan, V.H.; Rohatgi, R.; et al. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 2018, 37, 7452.
- D’Ambrogio, A.; Buratti, E.; Stuani, C.; Guarnaccia, C.; Romano, M.; Ayala, Y.M.; Baralle, F.E. Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic Acids Res. 2009, 37, 4116–4126.
- Buratti, E.; Brindisi, A.; Giombi, M.; Tisminetzky, S.; Ayala, Y.M.; Baralle, F.E. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 2005, 280, 37572–37584.
- Ayala, Y.M.; Pantano, S.; D’Ambrogio, A.; Buratti, E.; Brindisi, A.; Marchetti, C.; Romano, M.; Baralle, F.E. Human, Drosophila, and C.elegans TDP43: Nucleic acid binding properties and splicing regulatory function. J. Mol. Biol. 2005, 348, 575–588.
- Cartegni, L.; Maconi, M.; Morandi, E.; Cobianchi, F.; Riva, S.; Biamonti, G. hnRNP A1 selectively interacts through its Gly-rich domain with different RNA-binding proteins. J. Mol. Biol. 1996, 259, 337–348.
- Tziortzouda, P.; Van Den Bosch, L.; Hirth, F. Triad of TDP43 control in neurodegeneration: Autoregulation, localization and aggregation. Nat. Rev. Neurosci. 2021, 22, 197–208.
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672.
- Fratta, P.; Sivakumar, P.; Humphrey, J.; Lo, K.; Ricketts, T.; Oliveira, H.; Brito-Armas, J.M.; Kalmar, B.; Ule, A.; Yu, Y.; et al. Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J. 2018, 37, 8684.
- Bolognesi, B.; Faure, A.J.; Seuma, M.; Schmiedel, J.M.; Tartaglia, G.G.; Lehner, B. The mutational landscape of a prion-like domain. Nat. Commun. 2019, 10, 4162.
- Mann, J.R.; Donnelly, C.J. RNA modulates physiological and neuropathological protein phase transitions. Neuron 2021, 109, 2663–2681.
- Mompean, M.; Laurents, D.V. Intrinsically Disordered Domains, Amyloids and Protein Liquid Phases: Evolving Concepts and Open Questions. Protein Pept Lett. 2017, 24, 281–293.
- Lim, L.; Wei, Y.; Lu, Y.; Song, J. ALS-Causing Mutations Significantly Perturb the Self-Assembly and Interaction with Nucleic Acid of the Intrinsically Disordered Prion-Like Domain of TDP-43. PLoS Biol. 2016, 14, e1002338.
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468.
- Shiina, Y.; Arima, K.; Tabunoki, H.; Satoh, J. TDP-43 dimerizes in human cells in culture. Cell Mol. Neurobiol. 2010, 30, 641–652.
- Hallegger, M.; Chakrabarti, A.M.; Lee, F.C.Y.; Lee, B.L.; Amalietti, A.G.; Odeh, H.M.; Copley, K.E.; Rubien, J.D.; Portz, B.; Kuret, K.; et al. TDP-43 condensation properties specify its RNA-binding and regulatory repertoire. Cell 2021, 184, 4680–4696.
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293.
- Portz, B.; Lee, B.L.; Shorter, J. FUS and TDP-43 Phases in Health and Disease. Trends Biochem. Sci. 2021, 46, 550–563.
- Sternburg, E.L.; Gruijs da Silva, L.A.; Dormann, D. Post-translational modifications on RNA-binding proteins: Accelerators, brakes, or passengers in neurodegeneration? Trends Biochem. Sci. 2022, 47, 6–22.
- Igaz, L.M.; Kwong, L.K.; Lee, E.B.; Chen-Plotkin, A.; Swanson, E.; Unger, T.; Malunda, J.; Xu, Y.; Winton, M.J.; Trojanowski, J.Q.; et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Invest. 2011, 121, 726–738.
- Ayala, Y.M.; De Conti, L.; Avendano-Vazquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288.
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 2016, 139, 3187–3201.
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911.
- Smethurst, P.; Newcombe, J.; Troakes, C.; Simone, R.; Chen, Y.R.; Patani, R.; Sidle, K. In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol. Dis. 2016, 96, 236–247.
- Franceschini, A.; Baiardi, S.; Hughson, A.G.; McKenzie, N.; Moda, F.; Rossi, M.; Capellari, S.; Green, A.; Giaccone, G.; Caughey, B.; et al. High diagnostic value of second generation CSF RT-QuIC across the wide spectrum of CJD prions. Sci. Rep. 2017, 7, 10655.
- Orru, C.D.; Groveman, B.R.; Hughson, A.G.; Zanusso, G.; Coulthart, M.B.; Caughey, B. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. mBio 2015, 6.
- Fairfoul, G.; McGuire, L.I.; Pal, S.; Ironside, J.W.; Neumann, J.; Christie, S.; Joachim, C.; Esiri, M.; Evetts, S.G.; Rolinski, M.; et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 2016, 3, 812–818.
- Salvadores, N.; Shahnawaz, M.; Scarpini, E.; Tagliavini, F.; Soto, C. Detection of misfolded Abeta oligomers for sensitive biochemical diagnosis of Alzheimer’s disease. Cell Rep. 2014, 7, 261–268.
- Porta, S.; Xu, Y.; Restrepo, C.R.; Kwong, L.K.; Zhang, B.; Brown, H.J.; Lee, E.B.; Trojanowski, J.Q.; Lee, V.M. Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 2018, 9, 4220.
- Furukawa, Y.; Kaneko, K.; Watanabe, S.; Yamanaka, K.; Nukina, N. A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J. Biol. Chem. 2011, 286, 18664–18672.
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339.
- Scialo, C.; Tran, T.H.; Salzano, G.; Novi, G.; Caponnetto, C.; Chio, A.; Calvo, A.; Canosa, A.; Moda, F.; Caroppo, P.; et al. TDP-43 real-time quaking induced conversion reaction optimization and detection of seeding activity in CSF of amyotrophic lateral sclerosis and frontotemporal dementia patients. Brain Commun. 2020, 2, fcaa142.
- Beyer, L.; Gunther, R.; Koch, J.C.; Klebe, S.; Hagenacker, T.; Lingor, P.; Biesalski, A.S.; Hermann, A.; Nabers, A.; Gold, R.; et al. TDP-43 as structure-based biomarker in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2021, 8, 271–277.
- Mackenzie, I.R.; Neumann, M. Reappraisal of TDP-43 pathology in FTLD-U subtypes. Acta Neuropathol. 2017, 134, 79–96.
- Lee, E.B.; Porta, S.; Michael Baer, G.; Xu, Y.; Suh, E.; Kwong, L.K.; Elman, L.; Grossman, M.; Lee, V.M.; Irwin, D.J.; et al. Expansion of the classification of FTLD-TDP: Distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 2017, 134, 65–78.
- Chen, Z.; Ma, L. Grey matter volume changes over the whole brain in amyotrophic lateral sclerosis: A voxel-wise meta-analysis of voxel based morphometry studies. Amyotroph Lateral Scler 2010, 11, 549–554.
- Broe, M.; Hodges, J.R.; Schofield, E.; Shepherd, C.E.; Kril, J.J.; Halliday, G.M. Staging disease severity in pathologically confirmed cases of frontotemporal dementia. Neurology 2003, 60, 1005–1011.
- Chiang, C.H.; Grauffel, C.; Wu, L.S.; Kuo, P.H.; Doudeva, L.G.; Lim, C.; Shen, C.K.; Yuan, H.S. Structural analysis of disease-related TDP-43 D169G mutation: Linking enhanced stability and caspase cleavage efficiency to protein accumulation. Sci. Rep. 2016, 6, 21581.
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain 2019, 142, 1176–1194.
- Kametani, F.; Obi, T.; Shishido, T.; Akatsu, H.; Murayama, S.; Saito, Y.; Yoshida, M.; Hasegawa, M. Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci. Rep. 2016, 6, 23281.
- Kametani, F.; Nonaka, T.; Suzuki, T.; Arai, T.; Dohmae, N.; Akiyama, H.; Hasegawa, M. Identification of casein kinase-1 phosphorylation sites on TDP-43. Biochem. Biophys. Res. Commun. 2009, 382, 405–409.
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M.; et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70.
- Cracco, L.; Doud, E.H.; Hallinan, G.I.; Garringer, H.J.; Jacobsen, M.H.; Richardson, R.M.; Buratti, E.; Vidal, R.; Ghetti, B.; Newell, K.L. Distinguishing post-translational modifications in dominantly inherited frontotemporal dementias: FTLD-TDP Type A (GRN) vs Type B (C9orf72). Neuropathol. Appl. Neurobiol. 2022, 48, e12836.
- Laferriere, F.; Maniecka, Z.; Perez-Berlanga, M.; Hruska-Plochan, M.; Gilhespy, L.; Hock, E.M.; Wagner, U.; Afroz, T.; Boersema, P.J.; Barmettler, G.; et al. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat. Neurosci. 2019, 22, 65–77.
- Feneberg, E.; Charles, P.D.; Finelli, M.J.; Scott, C.; Kessler, B.M.; Fischer, R.; Ansorge, O.; Gray, E.; Talbot, K.; Turner, M.R. Detection and quantification of novel C-terminal TDP-43 fragments in ALS-TDP. Brain Pathol. 2021, 31, e12923.
- Pobran, T.D.; Forgrave, L.M.; Zheng, Y.Z.; Lim, J.G.K.; Mackenzie, I.R.A.; DeMarco, M.L. Detection and characterization of TDP-43 in human cells and tissues by multiple reaction monitoring mass spectrometry. Clin. Mass Spectrom. 2019, 14 Pt B, 66–73.
- Pobran, T.D.; Yang, D.; Mackenzie, I.R.A.; DeMarco, M.L. Aptamer-based enrichment of TDP-43 from human cells and tissues with quantification by HPLC-MS/MS. J. Neurosci. Methods 2021, 363, 109344.
- Donde, A.; Sun, M.; Ling, J.P.; Braunstein, K.E.; Pang, B.; Wen, X.; Cheng, X.; Chen, L.; Wong, P.C. Splicing repression is a major function of TDP-43 in motor neurons. Acta Neuropathol. 2019, 138, 813–826.
- Highley, J.R.; Kirby, J.; Jansweijer, J.A.; Webb, P.S.; Hewamadduma, C.A.; Heath, P.R.; Higginbottom, A.; Raman, R.; Ferraiuolo, L.; Cooper-Knock, J.; et al. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol. Appl. Neurobiol. 2014, 40, 670–685.
- Scaber, J.; Talbot, K. What is the role of TDP-43 in C9orf72-related amyotrophic lateral sclerosis and frontemporal dementia? Brain 2016, 139, 3057–3059.
- Vatsavayai, S.C.; Yoon, S.J.; Gardner, R.C.; Gendron, T.F.; Vargas, J.N.; Trujillo, A.; Pribadi, M.; Phillips, J.J.; Gaus, S.E.; Hixson, J.D.; et al. Timing and significance of pathological features in C9orf72 expansion-associated frontotemporal dementia. Brain 2016, 139, 3202–3216.
- Ling, J.P.; Pletnikova, O.; Troncoso, J.C.; Wong, P.C. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 2015, 349, 650–655.
- Melamed, Z.; Lopez-Erauskin, J.; Baughn, M.W.; Zhang, O.; Drenner, K.; Sun, Y.; Freyermuth, F.; McMahon, M.A.; Beccari, M.S.; Artates, J.W.; et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 2019, 22, 180–190.
- Brown, A.L.; Wilkins, O.G.; Keuss, M.J.; Hill, S.E.; Zanovello, M.; Lee, W.C.; Bampton, A.; Lee, F.C.Y.; Masino, L.; Qi, Y.A.; et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 2022, 603, 131–137.
- Prudencio, M.; Humphrey, J.; Pickles, S.; Brown, A.L.; Hill, S.E.; Kachergus, J.M.; Shi, J.; Heckman, M.G.; Spiegel, M.R.; Cook, C.; et al. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J. Clin. Invest. 2020, 130, 6080–6092.
- Theunissen, F.; Anderton, R.S.; Mastaglia, F.L.; Flynn, L.L.; Winter, S.J.; James, I.; Bedlack, R.; Hodgetts, S.; Fletcher, S.; Wilton, S.D.; et al. Novel STMN2 Variant Linked to Amyotrophic Lateral Sclerosis Risk and Clinical Phenotype. Front. Aging Neurosci. 2021, 13, 658226.
- Klim, J.R.; Williams, L.A.; Limone, F.; Guerra San Juan, I.; Davis-Dusenbery, B.N.; Mordes, D.A.; Burberry, A.; Steinbaugh, M.J.; Gamage, K.K.; Kirchner, R.; et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 2019, 22, 167–179.
- Ma, X.R.; Prudencio, M.; Koike, Y.; Vatsavayai, S.C.; Kim, G.; Harbinski, F.; Briner, A.; Rodriguez, C.M.; Guo, C.; Akiyama, T.; et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature 2022, 603, 124–130.
- Augustin, I.; Rosenmund, C.; Sudhof, T.C.; Brose, N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 1999, 400, 457–461.
- Deng, L.; Kaeser, P.S.; Xu, W.; Sudhof, T.C. RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron 2011, 69, 317–331.
- Lipstein, N.; Verhoeven-Duif, N.M.; Michelassi, F.E.; Calloway, N.; van Hasselt, P.M.; Pienkowska, K.; van Haaften, G.; van Haelst, M.M.; van Empelen, R.; Cuppen, I.; et al. Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder. J. Clin. Invest. 2017, 127, 1005–1018.
- Tan, H.H.G.; Westeneng, H.J.; van der Burgh, H.K.; van Es, M.A.; Bakker, L.A.; van Veenhuijzen, K.; van Eijk, K.R.; van Eijk, R.P.A.; Veldink, J.H.; van den Berg, L.H. The Distinct Traits of the UNC13A Polymorphism in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2020, 88, 796–806.
- Diekstra, F.P.; Van Deerlin, V.M.; van Swieten, J.C.; Al-Chalabi, A.; Ludolph, A.C.; Weishaupt, J.H.; Hardiman, O.; Landers, J.E.; Brown, R.H., Jr.; van Es, M.A.; et al. C9orf72 and UNC13A are shared risk loci for amyotrophic lateral sclerosis and frontotemporal dementia: A genome-wide meta-analysis. Ann. Neurol. 2014, 76, 120–133.
- Diekstra, F.P.; van Vught, P.W.; van Rheenen, W.; Koppers, M.; Pasterkamp, R.J.; van Es, M.A.; Schelhaas, H.J.; de Visser, M.; Robberecht, W.; Van Damme, P.; et al. UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 630-e3.
- van Es, M.A.; Veldink, J.H.; Saris, C.G.; Blauw, H.M.; van Vught, P.W.; Birve, A.; Lemmens, R.; Schelhaas, H.J.; Groen, E.J.; Huisman, M.H.; et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet. 2009, 41, 1083–1087.
- Hruska-Plochan, M.; Betz, K.M.; Ronchi, S.; Wiersma, V.I.; Maniecka, Z.; Hock, E.M.; Laferriere, F.; Sahadevan, S.; Hoop, V.; Delvendahl, I.; et al. Human neural networks with sparse TDP-43 pathology reveal NPTX2 misregulation in ALS/FTLD. bioRxiv 2021, 9, 12.
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; Konig, J.; Hortobagyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458.
- van der Ende, E.L.; Xiao, M.; Xu, D.; Poos, J.M.; Panman, J.L.; Jiskoot, L.C.; Meeter, L.H.; Dopper, E.G.; Papma, J.M.; Heller, C.; et al. Neuronal pentraxin 2: A synapse-derived CSF biomarker in genetic frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 2020, 91, 612–621.
- Cooper-Knock, J.; Green, C.; Altschuler, G.; Wei, W.; Bury, J.J.; Heath, P.R.; Wyles, M.; Gelsthorpe, C.; Highley, J.R.; Lorente-Pons, A.; et al. A data-driven approach links microglia to pathology and prognosis in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2017, 5, 23.
- Oeckl, P.; Weydt, P.; Thal, D.R.; Weishaupt, J.H.; Ludolph, A.C.; Otto, M. Proteomics in cerebrospinal fluid and spinal cord suggests UCHL1, MAP2 and GPNMB as biomarkers and underpins importance of transcriptional pathways in amyotrophic lateral sclerosis. Acta Neuropathol. 2020, 139, 119–134.
- Bian, Y.; Zheng, R.; Bayer, F.P.; Wong, C.; Chang, Y.C.; Meng, C.; Zolg, D.P.; Reinecke, M.; Zecha, J.; Wiechmann, S.; et al. Robust, reproducible and quantitative analysis of thousands of proteomes by micro-flow LC-MS/MS. Nat. Commun. 2020, 11, 157.
- Heywood, W.E.; Galimberti, D.; Bliss, E.; Sirka, E.; Paterson, R.W.; Magdalinou, N.K.; Carecchio, M.; Reid, E.; Heslegrave, A.; Fenoglio, C.; et al. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high-throughput multiplexed targeted proteomic assay. Mol. Neurodegener 2015, 10, 64.
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15.
- Umoh, M.E.; Dammer, E.B.; Dai, J.; Duong, D.M.; Lah, J.J.; Levey, A.I.; Gearing, M.; Glass, J.D.; Seyfried, N.T. A proteomic network approach across the ALS-FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol. Med. 2018, 10, 48–62.
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol. 2021, 141, 257–279.
- Paron, F.; Barattucci, S.; Cappelli, S.; Romano, M.; Berlingieri, C.; Stuani, C.; Laurents, D.; Mompean, M.; Buratti, E. Unraveling the toxic effects mediated by the neurodegenerative disease-associated S375G mutation of TDP-43 and its S375E phosphomimetic variant. J. Biol. Chem. 2022, 298, 102252.
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e618.
- Lucini, C.B.; Braun, R.J. Mitochondrion-Dependent Cell Death in TDP-43 Proteinopathies. Biomedicines 2021, 9, 376.
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Muller, K.; Marroquin, N.; Nordin, F.; Hubers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636.
- Brenner, D.; Sieverding, K.; Bruno, C.; Luningschror, P.; Buck, E.; Mungwa, S.; Fischer, L.; Brockmann, S.J.; Ulmer, J.; Bliederhauser, C.; et al. Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J. Exp. Med. 2019, 216, 267–278.
- Bevan-Jones, W.R.; Cope, T.E.; Jones, P.S.; Kaalund, S.S.; Passamonti, L.; Allinson, K.; Green, O.; Hong, Y.T.; Fryer, T.D.; Arnold, R.; et al. Neuroinflammation and protein aggregation co-localize across the frontotemporal dementia spectrum. Brain 2020, 143, 1010–1026.
- Steinacker, P.; Verde, F.; Fang, L.; Feneberg, E.; Oeckl, P.; Roeber, S.; Anderl-Straub, S.; Danek, A.; Diehl-Schmid, J.; Fassbender, K.; et al. Chitotriosidase (CHIT1) is increased in microglia and macrophages in spinal cord of amyotrophic lateral sclerosis and cerebrospinal fluid levels correlate with disease severity and progression. J. Neurol. Neurosurg. Psychiatry 2018, 89, 239–247.
- Thompson, A.G.; Gray, E.; Thezenas, M.L.; Charles, P.D.; Evetts, S.; Hu, M.T.; Talbot, K.; Fischer, R.; Kessler, B.M.; Turner, M.R. Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann. Neurol. 2018, 83, 258–268.
- Gille, B.; De Schaepdryver, M.; Dedeene, L.; Goossens, J.; Claeys, K.G.; Van Den Bosch, L.; Tournoy, J.; Van Damme, P.; Poesen, K. Inflammatory markers in cerebrospinal fluid: Independent prognostic biomarkers in amyotrophic lateral sclerosis? J. Neurol. Neurosurg. Psychiatry 2019, 90, 1338–1346.
- Abu-Rumeileh, S.; Steinacker, P.; Polischi, B.; Mammana, A.; Bartoletti-Stella, A.; Oeckl, P.; Baiardi, S.; Zenesini, C.; Huss, A.; Cortelli, P.; et al. CSF biomarkers of neuroinflammation in distinct forms and subtypes of neurodegenerative dementia. Alzheimers Res. Ther. 2019, 12, 2.
- Oeckl, P.; Weydt, P.; Steinacker, P.; Anderl-Straub, S.; Nordin, F.; Volk, A.E.; Diehl-Schmid, J.; Andersen, P.M.; Kornhuber, J.; Danek, A.; et al. Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J. Neurol. Neurosurg. Psychiatry 2019, 90, 4–10.
- Thompson, A.G.; Gray, E.; Bampton, A.; Raciborska, D.; Talbot, K.; Turner, M.R. CSF chitinase proteins in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1215–1220.
- Illan-Gala, I.; Alcolea, D.; Montal, V.; Dols-Icardo, O.; Munoz, L.; de Luna, N.; Turon-Sans, J.; Cortes-Vicente, E.; Sanchez-Saudinos, M.B.; Subirana, A.; et al. CSF sAPPbeta, YKL-40, and NfL along the ALS-FTD spectrum. Neurology 2018, 91, e1619–e1628.
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438.
- de Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2020.
- Del Campo, M.; Zetterberg, H.; Gandy, S.; Onyike, C.U.; Oliveira, F.; Udeh-Momoh, C.; Lleo, A.; Teunissen, C.E.; Pijnenburg, Y. New developments of biofluid-based biomarkers for routine diagnosis and disease trajectories in frontotemporal dementia. Alzheimers Dement. 2022, 18, 2292–22307.
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208.
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211.
- Chio, A.; Mazzini, L.; D’Alfonso, S.; Corrado, L.; Canosa, A.; Moglia, C.; Manera, U.; Bersano, E.; Brunetti, M.; Barberis, M.; et al. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology 2018, 91, e635–e642.
- Pasetto, L.; Grassano, M.; Pozzi, S.; Luotti, S.; Sammali, E.; Migazzi, A.; Basso, M.; Spagnolli, G.; Biasini, E.; Micotti, E.; et al. Defective cyclophilin A induces TDP-43 proteinopathy: Implications for amyotrophic lateral sclerosis and frontotemporal dementia. Brain 2021, 144, 3710–3726.
- Nardo, G.; Pozzi, S.; Pignataro, M.; Lauranzano, E.; Spano, G.; Garbelli, S.; Mantovani, S.; Marinou, K.; Papetti, L.; Monteforte, M.; et al. Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS ONE 2011, 6, e25545.
- Pasetto, L.; Callegaro, S.; Corbelli, A.; Fiordaliso, F.; Ferrara, D.; Brunelli, L.; Sestito, G.; Pastorelli, R.; Bianchi, E.; Cretich, M.; et al. Decoding distinctive features of plasma extracellular vesicles in amyotrophic lateral sclerosis. Mol. Neurodegener 2021, 16, 52.
- Lehmer, C.; Oeckl, P.; Weishaupt, J.H.; Volk, A.E.; Diehl-Schmid, J.; Schroeter, M.L.; Lauer, M.; Kornhuber, J.; Levin, J.; Fassbender, K.; et al. Poly-GP in cerebrospinal fluid links C9orf72-associated dipeptide repeat expression to the asymptomatic phase of ALS/FTD. EMBO Mol. Med. 2017, 9, 859–868.
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chio, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110.
- Winer, L.; Srinivasan, D.; Chun, S.; Lacomis, D.; Jaffa, M.; Fagan, A.; Holtzman, D.M.; Wancewicz, E.; Bennett, C.F.; Bowser, R.; et al. SOD1 in cerebral spinal fluid as a pharmacodynamic marker for antisense oligonucleotide therapy. JAMA Neurol. 2013, 70, 201–207.
- Eitan, C.; Hornstein, E. Vulnerability of microRNA biogenesis in FTD-ALS. Brain Res. 2016, 1647, 105–111.
- Gascon, E.; Gao, F.B. The emerging roles of microRNAs in the pathogenesis of frontotemporal dementia-amyotrophic lateral sclerosis (FTD-ALS) spectrum disorders. J. Neurogenet. 2014, 28, 30–40.
- Freischmidt, A.; Muller, K.; Zondler, L.; Weydt, P.; Volk, A.E.; Bozic, A.L.; Walter, M.; Bonin, M.; Mayer, B.; von Arnim, C.A.; et al. Serum microRNAs in patients with genetic amyotrophic lateral sclerosis and pre-manifest mutation carriers. Brain 2014, 137, 2938–2950.
- Kawahara, Y.; Mieda-Sato, A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352.
- Buratti, E.; De Conti, L.; Stuani, C.; Romano, M.; Baralle, M.; Baralle, F. Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J. 2010, 277, 2268–2281.
- Freischmidt, A.; Muller, K.; Ludolph, A.C.; Weishaupt, J.H. Systemic dysregulation of TDP-43 binding microRNAs in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2013, 1, 42.
- Dolinar, A.; Koritnik, B.; Glavac, D.; Ravnik-Glavac, M. Circular RNAs as Potential Blood Biomarkers in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2019, 56, 8052–8062.
- Gagliardi, S.; Zucca, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. Long non-coding and coding RNAs characterization in Peripheral Blood Mononuclear Cells and Spinal Cord from Amyotrophic Lateral Sclerosis patients. Sci. Rep. 2018, 8, 2378.
- Liu, W.; Wang, Z.; Liu, L.; Yang, Z.; Liu, S.; Ma, Z.; Liu, Y.; Ma, Y.; Zhang, L.; Zhang, X.; et al. LncRNA Malat1 inhibition of TDP43 cleavage suppresses IRF3-initiated antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2020, 117, 23695–23706.
- Matsukawa, K.; Kukharsky, M.S.; Park, S.K.; Park, S.; Watanabe, N.; Iwatsubo, T.; Hashimoto, T.; Liebman, S.W.; Shelkovnikova, T.A. Long non-coding RNA NEAT1_1 ameliorates TDP-43 toxicity in in vivo models of TDP-43 proteinopathy. RNA Biol. 2021, 18, 1546–1554.
This entry is offline, you can click here to edit this entry!