Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Taxanes are a class of diterpenes originally isolated from plants of the yew family (Taxaceae). An intriguing alternative to natural vesicles is represented by bioinspired extracellular vesicle (EV)-like nanoparticles (NPs), for example exosomes (EXOs) obtained from a serial extrusion process of a parent cell membrane suspension through decreasing pore size membranes or from hybridization of EXOs and liposome membranes.
- cancer
- plant derivatives
- extracellular vesicles
- cell-derived vesicles
- hybrid vesicles
- biomimetic nanoparticles
1. Introduction
Nowadays about 60% of the commonly used anticancer drugs were obtained from natural sources [1][2], either inspired by traditional medicine [3] or revealed by experimental findings [4]. Herbal medicines in the form of teas, tinctures, powders and poultices have been used for over 5000 years [5] in traditional medicines, still diffused in 88% of all countries, according to the WHO Global Centre for Traditional Medicine.
Medicinal plants are rich in diverse phytochemicals, such as flavonoids, terpenoids, alkaloids, saponins, lignins, coumarines, stilbenes, and taxanes [6][7], which are present in different parts of the plant, such as flowers, flower stigmas, pericarp, fruits, seeds, sprouts, roots, leaves, embryo, bark, and rhizomes [6]. Some of these compounds may exert anticancer effects through different mechanisms, for example through the inhibition of cancer-cell activating pathways, potentiation of DNA repair mechanisms, induction of antioxidant action, and stimulation of protective enzyme formation [6][8]. To identify such active compounds, a multistep process involving different fields of expertise is required [3][9].
Despite their beneficial potential, the use of phytochemicals for cancer treatment is frequently hampered by bioavailability issues, due to poor water solubility, high metabolization rate and low chemical stability [10][11][12][13][14], issues that may be overcome by including the phytochemical in a nanoformulation [15][16][17][18][19][20][21][22]. Among the most investigated nanovehicles are liposomes and lipid-based nanosystems [23][24][25][26][27][28], micelles [29][30][31][32][33], polymeric nanoparticles (NPs) [34][35][36], niosomes [13][37][38][39] and nanosponges [40][41][42][43][44][45]. Moreover, besides nanoparticles (NPs), nanosizing techniques leading to the formation of active nanocrystals have been employed to formulate camptothecin [46][47], curcumin [48], quercetin [49], and paclitaxel [50][51].
Recently, cell-derived vesicles have attracted a great deal of interest in the field of drug delivery. Extracellular vesicles (EVs) are a heterogeneous class of membrane-derived vesicles secreted by all cell types [52] which, by vehiculating proteins and genetic material, are involved in cell-to-cell communication [53]. Nevertheless, controversial data have been reported about the role of cancer cell-secreted EVs, either promoting tumor progression [54], or exerting an antitumor effect [55].
2. Taxane-Vehiculating Exosomes and Bioinspired Vesicles
The yew trees began to attract chemists’ attention in the mid-1800s, when a powder named taxine was isolated, but it was only in the early 1960s that the first structures of taxane diterpenes and pseudoalkaloid diterpenoids were elucidated [56]. Starting from the 1990s, three taxanes have been employed in the treatment of various cancers: paclitaxel (PTX), which was approved for medical use as Taxol® in 1993; docetaxel, a synthetic derivative of PTX [57] that entered the market in 1995 under the brand name Taxotere®; and the semi-synthetic cabazitaxel [56], marketed since 2010 under the trade name Jevtana®, which was demonstrated to be superior to PTX and docetaxel, due to the reduced affinity for the multidrug-resistant P-glycoprotein (P-gp) [58]. Taxanes exert anticancer activity by binding β-tubulin and promoting the formation of irreversible assemblies of microtubules, slowing or blocking mitosis and inducing apoptotic cell death [59]. Moreover, taxanes alter multiple cellular oncogenic processes, including angiogenesis, inflammatory response, and ROS production [57].
The clinical use of taxanes is hampered by some serious side effects, including myelosuppression, neuropathy, allergic reactions, and gastrointestinal toxicity, but also by the emergence of drug resistance that occurs according to different mechanisms, such as overexpression of efflux transporters, defective apoptotic machineries, alterations of drug targets, and barriers in drug transport [60]. Formulating taxanes in nanoparticulate systems, and in particular bioinspired-nanosystems, might help in overcoming these issues.
Several groups focused their research on taxane-vehiculating exosomes (EXOs), attempting to employ vesicles derived from different sources and either loading PTX in the parent cells or incorporating it into isolated vesicles by incubation, possibly aided by sonication. For example, Pascucci et al. [61] developed PTX-loaded EXOs starting from murine mesenchymal stem cells (MSCs) derived from bone marrow, selected for their easy isolation and in vitro expansion procedures and for their ability to migrate into the tumor mass after systemic injection [62][63]. Indeed, the authors of this research, after observing that PTX-primed MSCs exerted antitumoral effects on different types of tumor cells in vitro and in vivo [64], wondered whether PTX release from the cells was mediated by EVs. For the priming, MSCs were exposed to PTX for 24 h, and then the EVs were isolated from the cell medium by differential centrifugations. Transmission electron microscopy (TEM) observation of the isolated EVs revealed a vesicle population with size ranging from 10 to 150 nm, suggesting that the isolation procedure led to the selection of a subpopulation mainly composed of EXOs, and the presence of PTX in the EVs was assessed by HPLC analysis and Fourier-transform infrared (FTIR) spectroscopy. The isolated EVs exhibited an antitumor effect on human pancreatic adenocarcinoma cells by inducing up to 80% of tumor growth inhibition vs. controls.
In addition, Melzer et al. [65] selected MSCs (of human origin, in this case) as a source of EXOs, and primed them with PTX. In the nanoparticle tracking analysis (NTA), the isolated PTX-EXOs exhibited a negative zeta potential (−43 mV) and an average diameter of 204 nm, slightly larger than the one of the empty EXOs obtained from non-primed MSCs, while the presence of protein biomarkers on the vesicle surface was assessed using immunoblot analysis; moreover, the PTX-primed cells secreted higher amounts of EXOs, compared to non-primed cells. The concentration-dependent cytotoxicity of the formulation was evidenced by a fluoroscan assay performed on three human cancer cell lines (A549 lung cancer, SK-OV-3 ovarian cancer, and hybrid MDA-hyb1 breast cancer cells), while the administration of PTX-EXOs to breast tumor-bearing mice resulted in a 64% reduction in the tumor weight, measured after animal sacrifice.
With the aim of circumventing the multiple drug resistance limiting the efficacy of many chemotherapeutics, Kim and his group [66] loaded PTX into murine macrophage-derived EXOs. After isolating the vesicles using a commercial kit, PTX loading was achieved either by incubation, electroporation, or sonication. EXO-PTX obtained by sonication, examined by dynamic light scattering (DLS), exhibited higher mean hydrodynamic diameter (288 nm) and lower zeta potential (−14 mV), compared to the ones obtained by incubation and electroporation; the sonication procedure also led to the highest drug loading (DL = 28%, measured by HPLC), while not modifying the protein content, as confirmed by Western blot analysis. Atomic force microscopy (AFM) showed that the EXOs retained their round-shape morphology after PTX loading through the different procedures, and dialysis followed by HPLC analysis evidenced a three-hour burst release of PTX, followed by a sustained release. An evident accumulation of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindo-carbocyanine perchlorate (DiI)-labeled PTX-EXOs in a murine Lewis lung carcinoma cell subline (3LL-M27) was assessed by confocal imaging. Moreover, an MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazoliumbromide) assay evidenced that the PTX-EXOs exerted higher antitumor effect on resistant MDCK canine cancer cells expressing high levels of P-gp, compared to PTX alone. Ex vivo confocal images revealed that 98% of EXOs co-localized with the lung metastases which were excised from mice intranasally administered with PTX-EXOs, indicating the efficient in vivo targeting of the nanosystem; this accumulation also resulted in the reduced progression of pulmonary metastasis in treated mice, monitored through bioluminescence of the lung cancer cells used to create the cancer animal model.
In a following work [67], the authors optimized the PTX-EXO formulation by incorporating aminoethylanisamide-polyethylene glycol (AA-PEG) in the vesicle membranes, providing stealth properties [68] and the ability to target the sigma receptor overexpressed in lung cancer cells [69]. To prepare the targeted PTX-loaded EXOs (AA-PEG-EXO-PTX), AA-PEG was added to a mixture of EXOs and PTX, followed by sonication and incubation. The AA-PEG amount was optimized in order to achieve maximal DL, which resulted in about 33% (amount of the drug/amount of EXO proteins). Western blot analysis confirmed that the preparation procedure did not affect EXO protein content, while DLS analysis revealed increased average size and zeta potential of AA-PEG-EXO-PTX (304 nm and −4.4 mV), compared to plain EXOs and PTX-EXOs. DiI-labeled AA-PEG-EXO-PTX were uptaken by murine lung cancer cells more than the controls, evidence also confirmed by the ex vivo observation of the co-localization of AA-PEG-EXO-PTX with pulmonary metastases in lung cancer-bearing mice, with consequent increased therapeutic efficacy.
In addition, Wang and his group prepared PTX-loaded EXOs isolated from murine macrophages [70]. In particular, through sequential centrifugations, the EXOs were isolated from lipopolysaccharide-activated M1 macrophages, able to produce EXOs with documented antitumor activity [71]. PTX was loaded through sonication followed by incubation at 37 °C, to restore the EXO membranes, obtaining a DL of 19%. DLS measurements revealed an increased size of 173 nm for PTX-EXOs, compared to plain EXOs and sonicated EXOs, probably due to the partial adsorption of PTX to the exosomal membranes, while TEM observation showed that no morphological changes occurred after sonication and PTX loading. An MTT assay performed on 4T1 breast cancer cells evidenced an anti-proliferative effect of PTX-EXOs, also confirmed by a flow cytometry analysis detecting a higher number of apoptotic cells after treatment with PTX-EXOs, compared to the controls. An in vivo test on mice xenografted with 4T1 cells evidenced reduced tumor volume and prolonged survival in those animals receiving PTX-EXOs, compared to the controls, which were treated with plain EXOs and free PTX.
An alternative source of EXOs is represented by cow milk: indeed, Agrawal and coworkers proposed milk-derived EXOs as oral carriers of PTX [72]. EXOs were isolated from raw milk using a sequential centrifugation procedure [73], and PTX was loaded by incubation. PTX loading caused an increase in EXO size (from 75 to 108 nm), zeta potential (from −15 to −7 mV), and polydispersity index (PDI, from 0.15 to 0.19), determined by DLS; these changes might be due, as stated by the authors, to the partial absorption of PTX to the vesicle surface through hydrophobic interactions. Unfortunately, the DL, measured by ultra-performance liquid chromatography (UPLC), was just 8%. The stability of the formulation was confirmed by monitoring the particle size in simulated gastrointestinal fluids, with a PTX release of about 20% after 2 h, increasing to 40% after 4 h; importantly, the drug release was not influenced by the different pH of the media used in the assay. The PTX-EXOs yielded higher tumor-growth inhibition and lower systemic toxicity in lung cancer-bearing mice, compared to plain EXOs and free PTX.
Similarly, Kumar and his group developed PTX-loaded EXOs deriving from bovine milk [74] for oral delivery, but they also functionalized the vesicles with folic acid (FA), considering the overexpression of folic acid receptor in tumor cells [75]. After isolating the EXOs from raw milk through sequential centrifugations, the active was loaded by sonication (in this case not followed by incubation at 37 °C); FA was linked to the vesicle surface through EDC coupling. The final FA-PTX-EXOs exhibited, through DLS, an average size of 95 nm, negative zeta potential (−27 mV) and low PDI (0.158). Encapsulation efficiency (EE) and DL, determined by UV spectrophotometry, were 82% and 26%, respectively; scanning electron microscopy (SEM) evidenced the spherical morphology of the final vesicles. Interestingly, the authors froze and lyophilized the FA-PTX-EXOs, after the addition of trehalose as a cryoprotectant, and the reconstituted vesicles exhibited no significant changes in size and drug content. The formulation showed a burst release of PTX (25%) in PBS medium in the first hour, followed by a sustained release for up to 48 h. Confocal microscopy revealed that FA-functionalized EXOs encapsulating the fluorescent coumarin-6 internalized in MDA-MB-231 human breast cancer cells (overexpressing the FA receptor) and localized within the lysosomes. The MTT assay allowed the determination of FA-PTX-EXOs IC50, lower than the one of free PTX and PTX-EXOs; moreover, the annexin-V assay proved the apoptotic inductive effect of FA-PTX-EXOs, also accompanied by the reduction in cell migration ability, investigated using a transwell migration assay. Unfortunately, an in vivo assay proving the gastro-intestinal absorption of the nanosystem was not reported by the authors.
To target glioblastoma, Salarpour and her group isolated human glioblastoma-derived EXOs, using a commercial purification kit, and loaded them with PTX, either by incubation or sonication [76]. DLS measurements revealed that the PTX-EXOs had an average size of 63 nm and 86 nm by following the incubation and sonication procedures, respectively, with negative zeta potential (between −18 and −22 mV). TEM and SEM confirmed the spherical morphology of the vesicles and, using a commercial assay, the presence of the CD9 tetraspanin surface protein biomarker was confirmed. Unfortunately, PTX loading, determined by HPLC, resulted in only 0.74% for incubation and 0.92% for the sonication method. The MTT assay confirmed higher glioblastoma cell growth inhibition of PTX-EXOs, compared to free PTX and plain EXOs.
Differently from the abovementioned papers, some researchers have been dedicated to the development of taxane-loaded bio-inspired particles, mimicking the EVs in their structure and homing abilities. In these research works, various bioderived membranes, obtained either from healthy or from cancer cells, have been tested on several types of synthetic NPs.
A widely investigated strategy is to develop red blood cell (RBC)-mimicking nanosystems by covering different kinds of NPs with RBC membranes, providing the nanosystem with biocompatibility and protecting it from circulation clearance; moreover, RBCs are an abundant source of biomembranes and, lacking internal organelles, their workup is easy.
In this context, Su et al. [77] proposed polymeric NPs composed of a core of poly(caprolactone) (PCL) and encapsulating PTX, coated with RBC membranes. RBCs were isolated from heparinized mouse blood and membrane fragments were obtained after sequential centrifugations; then, RBC membranes were resuspended, sonicated and extruded through 400 nm and 200 nm polycarbonate filters. The core NPs were prepared through the nanoprecipitation method by adding dropwise a PCL and PTX acetone solution to an aqueous solution of Poloxamer 188, then the mixture was homogenized and centrifuged. The RBC membrane coating was applied by co-extrusion, obtaining the final RVNPs. DLS measurements evidenced increased average size after the coating (from 134 to 148 nm), while the negative zeta potential remained substantially unchanged; TEM micrographs confirmed the effective covering of the NPs with the membranes. The DL and EE of RVPNs, probably determined by HPLC (the analytical method combined with dialysis for determining the drug release profile), were 4.0% and 95.7%, respectively; the coated particles showed a slower release profile compared to the uncoated ones. The immune escape ability of the RVNPs was assessed in vitro by incubating the nanosystem with mouse macrophages and observing scarce internalization using confocal fluorescence laser scanning microscopy and flow cytometry. In vitro studies on murine breast cancer cells revealed an RVNPs internalization comparable to that of the uncovered NPs, and consequently negligibly impaired by the RBC membrane coating; interestingly, the authors observed that the co-administration of RVNPs with the iRGD peptide in SD rats led to remarkably enhanced PTX permanence in blood circulation.
In a following work, the same authors [78], to overcome the drawbacks related to their RBC-coated nanosystem, including the lack of targeting ability and the difficulty of PTX release from the NPs due to the presence of intact RBC membranes forming a barrier, integrated the near infra-red (NIR) laser-responsive 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR) into their RBC-mimetic NPs. Indeed, irradiation of an NIR photosensitizer with a laser of 650–900 nm might enhance tumor penetration with remote and precise control [79]. The RBC-coated NPs (PTX-PN@DiR-RV) were prepared by co-extrusion of the DiR-labeled RBC membranes with the polymeric core, which included also the thermosensitive phospholipid 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC): indeed, this lipid undergoes a thermal transition triggered by the DiR absorption of an NIR radiation and thermal energy release, leading to the destruction of the polymeric core and the consequent release of the loaded drug. DLS measurement of the final coated NPs revealed an average hydrodynamic diameter of 151 nm and a zeta potential of −14 mV; the DLs of DiR and PTX were 10.1% and 4.1%, respectively. After observing the enhanced PTX release in PBS upon NIR stimulation of the coated NPs, the authors evidenced that laser radiation enhanced the cellular uptake of fluorescently-labeled PTX in murine breast cancer cells treated with PTX-PN@DiR-RV, accompanied also by a reduced cell viability, determined by a sulforhodamine B staining assay. Ex vivo and in vivo studies on breast cancer-bearing mice revealed radiation-enhanced peritumoral accumulation of PTX-PN@DiR-RV, with PTX concentrations 2.1-, 2.4-, and 2.3-fold higher than those without irradiation at 4.5, 8, and 24 h post-injection, respectively, correlated with significant tumor-growth suppression.
To improve PTX loading, Zhai et al. [80] proposed biomimetic NPs composed of a core of hydrophobic PTX nanocrystals surrounded by an intermediate amphiphilic layer of polyethylene glycol (PEG)-conjugated PTX, covered with an erythrocyte-mimicking shell (EM). For the synthesis of PEG-PTX, whose structure was confirmed by 1H-NMR, a carboxylic moiety was introduced on PEG by a reaction with succinic anhydride and then activated by EDC, and the obtained molecule was reacted with PTX. To investigate the effect of different NPs shape, the authors prepared both rod-shape pure PTX NPs through the film dispersion method and subsequent incubation–sonication procedure, and spherical-shaped NPs using the emulsion-lyophilized crystallization method; then, these NPs were covered with the PTX-PEG layer using probe sonication followed by lyophilization, centrifugation and resuspension. To prepare the EM-coated NPs, PEGylated PTX NPs were mixed and sonicated with the vesicles obtained from murine RBCs through sequential centrifugations, sonication and extrusion. The EM-coating of rod-shaped NPs was not successful, as evidenced by TEM, while EM-coated spherical PEG-PTX NPs showed, through DLS, an average diameter of 327 nm, minimally larger than the uncoated NPs, and a slightly negative zeta potential. Sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE) analysis confirmed that the final coated NPs retained almost all membrane proteins, compared to the original erythrocyte membranes. PTX loading was over 60%, with slow release in sink conditions. The EM-coated NPs, after labeling of the NP core with the fluorescent DiR dye, exhibited higher uptake and antitumor effect in different cancer cell lines, compared to the uncoated NPs. In vivo imaging of breast cancer-bearing mice evidenced accumulation of the EM-coated NPs at the tumor site, and ex vivo observation of the dissected organs showed an inhibition of tumor growth, in addition to metastasis formation prevention (Figure 1).
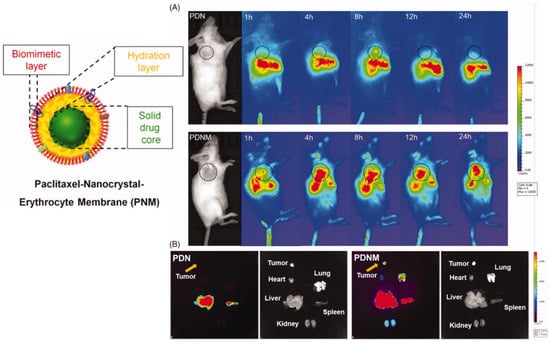
Figure 1. Scheme of EM-coated PEG-PTX NPs and in vivo NIR fluorescence imaging of DiR-labeled uncoated (PDN) and EM-coated (PDNM) PEG-PTX NPs. (A) In vivo imaging of 4T1 cancer-bearing BALB/c mice receiving a single injection of PDN or PDNM, respectively. The dashed black circles indicate the tumor burden. (B) Ex vivo imaging of tumor and organs excised from 4T1 tumor-bearing mice 24 h post injection of the two fluorescent formulations. Compared to PDN, the PDNM showed higher drug distributions at the tumor site. The yellow arrows point to the tumor tissue. Reproduced from [80] under the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/ accessed on 28 March 2023).
Song and coworkers [81] exploited murine RBC membranes for the development of pH-sensitive biomimetic NPs for the vehiculation of PTX. Using ultrasounds (US), the authors prepared a polymeric core composed of carboxymethylcellulose grafted with stearic acid through a carbodiimide crosslinking reaction, confirmed by 1H-NMR. The RBC membranes were isolated using sequential centrifugations, sonication and extrusion, and, to achieve cancer targeting ability, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE)-PEG-FA was inserted in the membranes by incubation. The thus-obtained vesicles were mixed with the core NPs and subjected to US, and PTX was loaded by incubation (EE = 11.5%), obtaining the final PTX-FRCS NPs. Distinct core–shell spherical structures were observed using TEM, with an average size of 227 nm and zeta potential of −14 mV. PTX-FRCS NPs possessed a protein profile similar to the one of the original erythrocyte membrane, as evidenced by the SDS-PAGE analysis, while fluorescence-activated cell sorting (FACS) analysis demonstrated that the RBC coating prevented PTX-FRCS NPs uptake by murine macrophages. The pH sensitivity was assessed by observing increased PTX release under acidic conditions, simulating the tumor microenvironment, and confocal images highlighted the PTX-FRCS NPs internalization in different cancer cell lines. After observing the in vivo accumulation of PTX-FRCS NPs in mouse tumor xenografts, the authors demonstrated that the administration of PTX-FRCS NPs to mice xenografted with liver cancer, lung cancer and melanoma resulted in suppression of tumor growth.
To target malignant melanoma, Cao et al. [82] proposed PTX loaded-albumin NPs coated with murine macrophage membranes, based on the evidence that macrophage coating enhances tumor targeting [83]. Cell membranes were isolated using hypotonic lysis, followed by mechanical membrane fragmentation, differential centrifugation and extrusion, while albumin NPs were prepared using a nanoprecipitation process. The final membrane-coated albumin NPs (PTX-RANPS), obtained by co-extrusion of the two components, exhibited a hydrodynamic diameter of 189 nm, which is between the size of membrane vesicles and uncoated albumin NPs, and negative zeta potential, as measured by DLS. The effective coating was proved by TEM observation, and PTX EE, determined by LC-MS/MS, was 83.4%. Flow cytometry and confocal laser scanning microscopy performed on different cancer cell lines revealed a significantly higher uptake of PTX-RANPS, compared to the uncoated albumin NPs, possibly following a lysosomal pathway after internalization; moreover, PTX-RANPS exerted higher cell-viability reduction, compared to the controls. Ex vivo fluorescence observation proved tumor-specific distribution and prolonged circulation time of PTX-RANPS in melanoma-bearing mice, accompanied also by a reduction in tumor volume, although not significant if compared to the control animals.
Several research groups, investigating novel effective bioinspired nanosystems for PTX delivery, decided to exploit cancer cell membranes to camouflage artificial NPs. Indeed, cancer cells feature immune escape and homing abilities [84], and are ideal cell membrane sources, since they are easy to culture in vitro.
In this context, to improve the therapeutic effect against cervical cancer, Xu et al. [85] developed cancer cell membrane-camouflaged NPs for the simultaneous delivery of PTX and an siRNA targeting the oncogene E7, which is possibly associated with PTX resistance. In particular, the authors prepared a poly(lactic-co-glycolic) (PLGA) core encapsulating PTX and siRNA, using the double-emulsion solvent evaporation technique, and human cervical-cancer-cell membranes were isolated by sequential centrifugations and extrusion; the final Si/PNPs@HeLa were obtained by co-extrusion. DLS measurements revealed that the membrane coating led to a decrease in zeta potential, from −14 to −30 mV, and a size increase of about 15 nm, while TEM micrographs evidenced a core-shell structure with a spherical shape. The EE of PTX and siRNA, determined by HPLC, were 90.2% and 88.4%, respectively, and gel electrophoresis followed by Coomassie blue staining confirmed that the final NPs retained the membrane proteins characterizing HeLa cells. Quantitative reverse transcription (qRT-PCR) analysis showed a 75% Si/PNPs@HeLa knockdown effect on the E7 oncogene after 48 h, significantly higher than that of the uncoated NPs. In vivo biodistribution using NIR-fluorescence whole body imaging performed on HeLa tumor-bearing mice receiving labeled Si/PNPs@HeLa highlighted increased fluorescence signals in the tumors and reduced non-specific accumulation in healthy organs, compared to uncoated NPs, and the authors confirmed the synergistic action of siRNA-E7 and PTX by observing the smallest tumor volume and the largest necrotic area in mice treated with Si/PNPs@HeLa, compared to the controls.
Similarly, to obtain a targeted nanosystem for treating osteosarcoma, Cai et al. [86] coated PLGA NPs, loaded with PTX, with hybrid membranes obtained from human osteosarcoma (143B) and murine macrophage (RAW264) cell membranes, thus combining the tumor homing properties of the cancer cells with the tumor-escape abilities of macrophages. The PTX-loaded PLGA NPs were prepared using the nano-precipitation method, while cell membranes were isolated by gradient centrifugation and sonication of the original cells [87]. The hybrid membranes were prepared by ultrasonic treatment of a 1:1 dispersion of the two types of membranes, and the final PTX-PLGA@[143B-RAW] NPs were obtained by adding the PTX-loaded PLGA NPs to the hybrid membrane suspension, with ultrasonic treatment selected as the best method, compared to co-extrusion, since it produced NPs with more reproducible sizes and higher membrane protein content. The final NPs had a spherical morphology and a core-shell structure, as evidenced by TEM, with a diameter of about 220 nm, determined by DLS. PTX EE and DL were 64.9% and 4.2%, respectively, and PTX release, evaluated in a PBS buffer, was pH-dependent, increasing at the low pH typical of the tumor microenvironment. In vitro internalization tests on osteosarcoma cells evidenced higher uptake and cytotoxicity of the hybrid membrane-coated NPs; moreover, using TNF-α stimulated HUVEC cells to mimic the inflammatory environment, and the authors proved that the macrophage cell membrane coating could improve the chemotactic effect of the NPs on the inflammatory environment. Then, using a live imaging system, a higher accumulation of the PLGA@[143B-RAW] NPs was observed, and PTX-PLGA@[143B-RAW] NPs induced a higher decrease in tumor volume in osteosarcoma-bearing mice, compared to the controls.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics15051445
References
- Babaei, G.; Aliarab, A.; Abroon, S.; Rasmi, Y.; Aziz, S.G.G. Application of sesquiterpene lactone: A new promising way for cancer therapy based on anticancer activity. Biomed. Pharmacother. 2018, 106, 239–246.
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803.
- Fridlender, M.; Kapulnik, Y.; Koltai, H. Plant derived substances with anti-cancer activity: From folklore to practice. Front. Plant. Sci. 2015, 6, 799.
- Lopes, C.M.; Dourado, A.; Oliveira, R. Phytotherapy and Nutritional Supplements on Breast Cancer. Biomed. Res. Int. 2017, 2017, 7207983.
- Ioele, G.; Chieffallo, M.; Occhiuzzi, M.A.; De Luca, M.; Garofalo, A.; Ragno, G.; Grande, F. Anticancer Drugs: Recent Strategies to Improve Stability Profile, Pharmacokinetic and Pharmacodynamic Properties. Molecules 2022, 27, 5436.
- Iqbal, J.; Abbasi, B.A.; Mahmood, T.; Kanwal, S.; Ali, B.; Shah, S.A.; Khalil, A.T. Plant-derived anticancer agents: A green anticancer approach. Asia. Pac. J. Trop. Biomed. 2017, 7, 1129–1150.
- Jahan, I.; Onay, A. Potentials of plant-based substance to inhabit and probable cure for the COVID-19. Turk. J. Biol. 2020, 44, 228–241.
- Tariq, A.; Sadia, S.; Pan, K.W.; Ullah, I.; Mussarat, S.; Sun, F.; Abiodun, O.O.; Batbaatar, A.; Li, Z.L.; Song, D.G.; et al. A systematic review on ethnomedicines of anticancer plants. Phytother. Res. 2017, 31, 202–264.
- Choudhari, A.S.; Mandave, P.C.; Deshpande, M.; Ranjekar, P.; Prakash, O. Phytochemicals in Cancer Treatment: From Preclinical Studies to Clinical Practice. Front. Pharmacol. 2020, 10, 1614.
- Gao, S.; Basu, S.; Yang, Z.; Deb, A.; Hu, M. Bioavailability Challenges Associated with Development of Saponins as Therapeutic and Chemopreventive Agents. Curr. Drug Targets 2012, 13, 1885–1899.
- Thilakarathna, S.H.; Rupasinghe, H.P.V. Flavonoid Bioavailability and Attempts for Bioavailability Enhancement. Nutrients 2013, 5, 3367–3387.
- Aqil, F.; Munagala, R.; Jeyabalan, J.; Vadhanam, M.V. Bioavailability of phytochemicals and its enhancement by drug delivery systems. Cancer Lett. 2013, 334, 133–141.
- Schlich, M.; Lai, F.; Pireddu, R.; Pini, E.; Ailuno, G.; Fadda, A.M.; Valenti, D.; Sinico, C. Resveratrol proniosomes as a convenient nanoingredient for functional food. Food Chem. 2020, 310, 125950.
- Shen, Y.X.; Zhang, N.; Tian, J.L.; Xin, G.; Liu, L.; Sun, X.Y.; Li, B. Advanced approaches for improving bioavailability and controlled release of anthocyanins. J. Control. Release 2022, 341, 285–299.
- Valdes, K.; Morales, J.; Rodriguez, L.; Gunther, G. Potential use of nanocarriers with pentacyclic triterpenes in cancer treatments. Nanomedicine 2016, 12, 3139–3156.
- Bilia, A.R.; Piazzini, V.; Asprea, M.; Risaliti, L.; Vanti, G.; Bergonzi, M.C. Plants Extracts Loaded in Nanocarriers: An Emergent Formulating Approach. Nat. Prod. Commun. 2018, 13, 1157–1160.
- Lai, F.; Schlich, M.; Pireddu, R.; Fadda, A.M.; Sinico, C. Nanocrystals as Effective Delivery Systems of Poorly Water-soluble Natural Molecules. Curr. Med. Chem. 2019, 26, 4657–4680.
- Lin, M.H.; Hung, C.F.; Hsu, C.Y.; Lin, Z.C.; Fang, J.Y. Prodrugs in combination with nanocarriers as a strategy for promoting antitumoral efficiency. Future Med. Chem. 2019, 11, 2131–2150.
- Bilia, A.R.; Piazzini, V.; Risaliti, L.; Vanti, G.; Casamonti, M.; Wang, M.; Bergonzi, M.C. Nanocarriers: A Successful Tool to Increase Solubility, Stability and Optimise Bioefficacy of Natural Constituents. Curr. Med. Chem. 2019, 26, 4631–4656.
- Vanti, G. Recent strategies in nanodelivery systems for natural products: A review. Environ. Chem. Lett. 2021, 19, 4311–4326.
- Truzzi, E.; Rustichelli, C.; de Oliveira, E.R.; Ferraro, L.; Maretti, E.; Graziani, D.; Botti, G.; Beggiato, S.; Iannuccelli, V.; Lima, E.M.; et al. Nasal biocompatible powder of Geraniol oil complexed with cyclodextrins for neurodegenerative diseases: Physicochemical characterization and in vivo evidences of nose to brain delivery. J. Control. Release 2021, 335, 191–202.
- Vieira, I.R.S.; Conte, C.A. Nano-delivery systems for food bioactive compounds in cancer: Prevention, therapy, and clinical applications. Crit. Rev. Food Sci. Nutr. 2022, 1–26.
- Goniotaki, M.; Hatziantoniou, S.; Dimas, K.; Wagner, M.; Demetzos, C. Encapsulation of naturally occurring flavonoids into liposomes: Physicochemical properties and biological activity against human cancer cell lines. J. Pharm. Pharmacol. 2004, 56, 1217–1224.
- Mignet, N.; Seguin, J.; Romano, M.R.; Brulle, L.; Touil, Y.S.; Scherman, D.; Bessodes, M.; Chabot, G.G. Development of a liposomal formulation of the natural flavonoid fisetin. Int. J. Pharm. 2012, 423, 69–76.
- Shehata, E.M.M.; Gowayed, M.A.; El-Ganainy, S.O.; Sheta, E.; Elnaggar, Y.S.R.; Abdallah, O.Y. Pectin coated nanostructured lipid carriers for targeted piperine delivery to hepatocellular carcinoma. Int. J. Pharm. 2022, 619, 121712.
- Jampilek, J.; Kralova, K. Anticancer Applications of Essential Oils Formulated into Lipid-Based Delivery Nanosystems. Pharmaceutics 2022, 14, 2681.
- Jing, D.D.; Wu, W.; Chen, X.Z.; Xiao, H.W.; Zhang, Z.H.; Chen, F.X.; Zhang, Z.C.; Liu, J.X.; Shao, Z.W.; Pu, F.F. Quercetin encapsulated in folic acid-modified liposomes is therapeutic against osteosarcoma by non-covalent binding to the JH2 domain of JAK2 Via the JAK2-STAT3-PDL1. Pharmacol. Res. 2022, 182, 106287.
- Demirbolat, G.M.; Erdogan, O.; Coskun, G.P.; Cevik, O. PEG4000 modified liposomes enhance the solubility of quercetin and improve the liposome functionality: In vitro characterization and the cellular efficacy. Turk. J. Chem. 2022, 46, 1011–1023.
- Zhao, H.; Wang, Y.L.; Peng, J.R.; Zhang, L.; Qu, Y.; Chu, B.Y.; Dong, M.L.; Tan, L.W.; Qian, Z.Y. Biodegradable Self-Assembled Micelles Based on MPEG-PTMC Copolymers: An Ideal Drug Delivery System for Vincristine. J. Biomed. Nanotechnol. 2017, 13, 427–436.
- El-Far, S.W.; Helmy, M.W.; Khattab, S.N.; Bekhit, A.A.; Hussein, A.A.; Elzoghby, A.O. Phytosomal bilayer-enveloped casein micelles for codelivery of monascus yellow pigments and resveratrol to breast cancer. Nanomedicine 2018, 13, 481–499.
- Ghalehkhondabi, V.; Soleymani, M.; Fazlali, A. Folate-targeted nanomicelles containing silibinin as an active drug delivery system for liver cancer therapy. J. Drug Deliv. Sci. Technol. 2021, 61, 102157.
- Chen, C.; Du, S.Y.; Zhong, W.; Liu, K.G.; Qu, L.H.; Chu, F.Y.; Yang, J.J.; Han, X. Accurate delivery of pristimerin and paclitaxel by folic acid-linked nano-micelles for enhancing chemosensitivity in cancer therapy. Nano Converg. 2022, 9, 52.
- Tan, X.R.; Feng, K.K.; Le, J.Q.; Shen, J.W.; Shao, J.W. Self-assembled micelles of the natural medicine ginsenosides for cancer metastasis therapy. J. Ind. Eng. Chem. 2022, 116, 303–309.
- Abdifetah, O.; Na-Bangchang, K. Pharmacokinetic studies of nanoparticles as a delivery system for conventional drugs and herb-derived compounds for cancer therapy: A systematic review. Int. J. Nanomed. 2019, 14, 5659–5677.
- Mughees, M.; Wajid, S.; Samim, M. Cytotoxic potential of Artemisia absinthium extract loaded polymeric nanoparticles against breast cancer cells: Insight into the protein targets. Int. J. Pharm. 2020, 586, 119583.
- Mughees, M.; Wajid, S. Herbal Based Polymeric Nanoparticles as a Therapeutic Remedy for Breast Cancer. Anti-Cancer Agents Med. Chem. 2021, 21, 433–444.
- Alemi, A.; Farrokhifar, M.; Zare-Zardini, H.; Karamallah, M.H. A Comparison between the Anticancer Activities of Free Paclitaxel and Paclitaxel-Loaded Niosome Nanoparticles on Human Acute Lymphoblastic Leukemia Cell Line Nalm-6. Iran. J. Pediatr. Hematol. Oncol. 2018, 8, 153–160.
- Pourmoghadasiyan, B.; Tavakkoli, F.; Beram, F.M.; Badmasti, F.; Mirzaie, A.; Kazempour, R.; Rahimi, S.; Larijani, S.F.; Hejabi, F.; Sedaghatnia, K. Nanosized paclitaxel-loaded niosomes: Formulation, in vitro cytotoxicity, and apoptosis gene expression in breast cancer cell lines. Mol. Biol. Rep. 2022, 49, 3597–3608.
- Mehrabi, M.R.; Shokrgozar, M.A.; Toliyat, T.; Shirzad, M.; Izadyari, A.; Mofrad, L.Z.; Chiani, M.; Akbarzadeh, A. Enhanced Therapeutic Efficacy of Vincristine Sulfate for Lymphoma Using Niosome-Based Drug Delivery. Jundish. J. Nat. Pharm. Prod. 2020, 15, e82793.
- Sundararajan, M.; Thomas, P.A.; Venkadeswaran, K.; Jeganathan, K.; Geraldine, P. Synthesis and Characterization of Chrysin-Loaded beta-Cyclodextrin-Based Nanosponges to Enhance In-Vitro Solubility, Photostability, Drug Release, Antioxidant Effects and Antitumorous Efficacy. J. Nanosci. Nanotechnol. 2017, 17, 8742–8751.
- Rafati, N.; Zarrabi, A.; Caldera, F.; Trotta, F.; Ghias, N. Pyromellitic dianhydride crosslinked cyclodextrin nanosponges for curcumin controlled release; formulation, physicochemical characterization and cytotoxicity investigations. J. Microencapsul. 2019, 36, 715–727.
- Clemente, N.; Argenziano, M.; Gigliotti, C.L.; Ferrara, B.; Boggio, E.; Chiocchetti, A.; Caldera, F.; Trotta, F.; Benetti, E.; Annaratone, L.; et al. Paclitaxel-Loaded Nanosponges Inhibit Growth and Angiogenesis in Melanoma Cell Models. Front. Pharmacol. 2019, 10, 776.
- Hariri, G.; Edwards, A.D.; Merrill, T.B.; Greenbaum, J.M.; van der Ende, A.E.; Harth, E. Sequential Targeted Delivery of Paclitaxel and Camptothecin Using a Cross-Linked “Nanosponge” Network for Lung Cancer Chemotherapy. Mol. Pharm. 2014, 11, 265–275.
- Gigliotti, C.L.; Ferrara, B.; Occhipinti, S.; Boggio, E.; Barrera, G.; Pizzimenti, S.; Giovarelli, M.; Fantozzi, R.; Chiocchetti, A.; Argenziano, M.; et al. Enhanced cytotoxic effect of camptothecin nanosponges in anaplastic thyroid cancer cells in vitro and in vivo on orthotopic xenograft tumors. Drug Deliv. 2017, 24, 670–680.
- Allahyari, S.; Zahednezhad, F.; Khatami, M.; Hashemzadeh, N.; Zakeri-Milani, P.; Trotta, F. Cyclodextrin nanosponges as potential anticancer drug delivery systems to be introduced into the market, compared with liposomes. J. Drug Deliv. Sci. Technol. 2022, 67, 102931.
- Zhang, H.; Hollis, C.P.; Zhang, Q.; Li, T.L. Preparation and antitumor study of camptothecin nanocrystals. Int. J. Pharm. 2011, 415, 293–300.
- Wang, J.H.; Muhammad, N.; Li, T.T.; Wang, H.; Liu, Y.J.; Liu, B.N.; Zhan, H.L. Hyaluronic Acid-Coated Camptothecin Nanocrystals for Targeted Drug Delivery to Enhance Anticancer Efficacy. Mol. Pharm. 2020, 17, 2411–2425.
- Ji, P.; Wang, L.; Chen, Y.W.; Wang, S.Q.; Wu, Z.H.; Qi, X.L. Hyaluronic acid hydrophilic surface rehabilitating curcumin nanocrystals for targeted breast cancer treatment with prolonged biodistribution. Biomater. Sci. 2020, 8, 462–472.
- Manca, M.L.; Lai, F.; Pireddu, R.; Valenti, D.; Schlich, M.; Pini, E.; Ailuno, G.; Fadda, A.M.; Sinico, C. Impact of nanosizing on dermal delivery and antioxidant activity of quercetin nanocrystals. J. Drug Deliv. Sci. Technol. 2020, 55, 101482.
- Han, S.D.; Li, X.P.; Zhou, C.H.; Hu, X.P.; Zhou, Y.H.; Jin, Y.; Liu, Q.; Wang, L.Q.; Li, X.R.; Liu, Y. Further Enhancement in Intestinal Absorption of Paclitaxel by Using Transferrin-Modified Paclitaxel Nanocrystals. ACS Appl. Biomater. 2020, 3, 4684–4695.
- Sreeharsha, N.; Prasanthi, S.; Mahalakshmi, S.; Goudanavar, P.S.; Naveen, N.R.; Gowthami, B.; Fattepur, S.; Meravanige, G.; Asdaq, S.M.B.; Anwer, M.K.; et al. Enhancement of Anti-Tumoral Properties of Paclitaxel Nano-Crystals by Conjugation of Folic Acid to Pluronic F127: Formulation Optimization, In vitro and In vivo Study. Molecules 2022, 27, 7914.
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289.
- Ailuno, G.; Baldassari, S.; Lai, F.; Florio, T.; Caviglioli, G. Exosomes and Extracellular Vesicles as Emerging Theranostic Platforms in Cancer Research. Cells 2020, 9, 2569.
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFrvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, U619–U624.
- Ristorcelli, E.; Beraud, E.; Verrando, P.; Villard, C.; Lafitte, D.; Sbarra, V.; Lombardo, D.; Verine, A. Human tumor nanoparticles induce apoptosis of pancreatic cancer cells. FASEB J. 2008, 22, 3358–3369.
- Lange, B.M.; Conner, C.F. Taxanes and taxoids of the genus Taxus-A comprehensive inventory of chemical diversity. Phytochemistry 2021, 190, 112829.
- Mosca, L.; Ilari, A.; Fazi, F.; Assaraf, Y.G.; Colotti, G. Taxanes in cancer treatment: Activity, chemoresistance and its overcoming. Drug Resist. Updates 2021, 54, 100742.
- Paller, C.J.; Antonarakis, E.S. Cabazitaxel: A novel second-line treatment for metastatic castration-resistant prostate cancer. Drug Des. Dev. Ther. 2011, 5, 117–124.
- Yang, C.P.H.; Horwitz, S.B. Taxol((R)): The First Microtubule Stabilizing Agent. Int. J. Mol. Sci. 2017, 18, 1733.
- Wang, S.P.; Qiu, J.G.; Shi, Z.; Wang, Y.T.; Chen, M.W. Nanoscale drug delivery for taxanes based on the mechanism of multidrug resistance of cancer. Biotechnol. Adv. 2015, 33, 224–241.
- Pascucci, L.; Cocce, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Vigano, L.; Locatelli, A.; Sisto, F.; Doglia, S.M.; et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control. Release 2014, 192, 262–270.
- Reagan, M.R.; Kaplan, D.L. Concise Review: Mesenchymal Stem Cell Tumor-Homing: Detection Methods in Disease Model Systems. Stem Cells 2011, 29, 920–927.
- Xie, M.Y.; Tao, L.; Zhang, Z.Q.; Wang, W. Mesenchymal Stem Cells Mediated Drug Delivery in Tumor-Targeted Therapy. Curr. Drug Del. 2021, 18, 864–879.
- Pessina, A.; Bonomi, A.; Cocce, V.; Invernici, G.; Navone, S.; Cavicchini, L.; Sisto, F.; Ferrari, M.; Vigano, L.; Locatelli, A.; et al. Mesenchymal Stromal Cells Primed with Paclitaxel Provide a New Approach for Cancer Therapy. PLoS ONE 2011, 6, e28321.
- Melzer, C.; Rehn, V.; Yang, Y.Y.; Bahre, H.; von der Ohe, J.; Hass, R. Taxol-Loaded MSC-Derived Exosomes Provide a Therapeutic Vehicle to Target Metastatic Breast Cancer and Other Carcinoma Cells. Cancers 2019, 11, 798.
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 655–664.
- Kim, M.S.; Haney, M.J.; Zhao, Y.L.; Yuan, D.F.; Deygen, I.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 195–204.
- Kooijmans, S.A.A.; Fliervoet, L.A.L.; van der Meel, R.; Fens, M.; Heijnen, H.F.G.; Henegouwen, P.; Vader, P.; Schiffelers, R.M. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Control. Release 2016, 224, 77–85.
- van Waarde, A.; Rybczynska, A.A.; Ramakrishnan, N.K.; Ishiwata, K.; Elsinga, P.H.; Dierckx, R. Potential applications for sigma receptor ligands in cancer diagnosis and therapy. BBA Biomembr. 2015, 1848, 2703–2714.
- Wang, P.P.; Wang, H.H.; Huang, Q.Q.; Peng, C.; Yao, L.; Chen, H.; Qiu, Z.; Wu, Y.F.; Wang, L.; Chen, W.D. Exosomes from M1-Polarized Macrophages Enhance Paclitaxel Antitumor Activity by Activating Macrophages-Mediated Inflammation. Theranostics 2019, 9, 1714–1727.
- Tang, Z.L.; Tang, C.Y.; Sun, C.; Ying, X.J.; Shen, R.L. M1 macrophage-derived exosomes synergistically enhance the anti-bladder cancer effect of gemcitabine. Aging 2022, 14, 7364–7377.
- Agrawal, A.K.; Aqil, F.; Jeyabalan, J.; Spencer, W.A.; Beck, J.; Gachuki, B.W.; Alhakeem, S.S.; Oben, K.; Munagala, R.; Bondada, S.; et al. Milk-derived exosomes for oral delivery of paclitaxel. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1627–1636.
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Gupta, R.C. Bovine milk-derived exosomes for drug delivery. Cancer Lett. 2016, 371, 48–61.
- Kumar, D.N.; Chaudhuri, A.; Dehari, D.; Shekher, A.; Gupta, S.C.; Majumdar, S.; Krishnamurthy, S.; Singh, S.; Kumar, D.; Agrawal, A.K. Combination Therapy Comprising Paclitaxel and 5-Fluorouracil by Using Folic Acid Functionalized Bovine Milk Exosomes Improves the Therapeutic Efficacy against Breast Cancer. Life 2022, 12, 1143.
- Assaraf, Y.G.; Leamon, C.P.; Reddy, J.A. The folate receptor as a rational therapeutic target for personalized cancer treatment. Drug Resist. Updates 2014, 17, 89–95.
- Salarpour, S.; Forootanfar, H.; Pournamdari, M.; Ahmadi-Zeidabadi, M.; Esmaeeli, M.; Pardakhty, A. Paclitaxel incorporated exosomes derived from glioblastoma cells: Comparative study of two loading techniques. Daru 2019, 27, 533–539.
- Su, J.H.; Sun, H.P.; Meng, Q.S.; Yin, Q.; Tang, S.; Zhang, P.C.; Chen, Y.; Zhang, Z.W.; Yu, H.J.; Li, Y.P. Long Circulation Red-Blood-Cell-Mimetic Nanoparticles with Peptide-Enhanced Tumor Penetration for Simultaneously Inhibiting Growth and Lung Metastasis of Breast Cancer. Adv. Funct. Mater. 2016, 26, 1243–1252.
- Su, J.H.; Sun, H.P.; Meng, Q.S.; Yin, Q.; Zhang, P.C.; Zhang, Z.W.; Yu, H.J.; Li, Y.P. Bioinspired Nanoparticles with NIR-Controlled Drug Release for Synergetic Chemophotothermal Therapy of Metastatic Breast Cancer. Adv. Funct. Mater. 2016, 26, 7495–7506.
- Luo, S.L.; Zhang, E.L.; Su, Y.P.; Cheng, T.M.; Shi, C.M. A review of NIR dyes in cancer targeting and imaging. Biomaterials 2011, 32, 7127–7138.
- Zhai, Z.; Xu, P.C.; Yao, J.; Li, R.D.; Gong, L.D.; Yin, Y.X.; Lin, Z.Q. Erythrocyte-mimicking paclitaxel nanoparticles for improving biodistributions of hydrophobic drugs to enhance antitumor efficacy. Drug Deliv. 2020, 27, 387–399.
- Song, M.; Dong, S.; An, X.; Zhang, W.; Shen, N.; Li, Y.; Guo, C.; Liu, C.; Li, X.; Chen, S. Erythrocyte-biomimetic nanosystems to improve antitumor effects of paclitaxel on epithelial cancers. J. Control. Release 2022, 345, 744–754.
- Cao, X.; Tan, T.F.; Zhu, D.C.; Yu, H.X.; Liu, Y.R.; Zhou, H.Y.; Jin, Y.; Xia, Q. Paclitaxel-Loaded Macrophage Membrane Camouflaged Albumin Nanoparticles for Targeted Cancer Therapy. Int. J. Nanomed. 2020, 15, 1915–1928.
- Khatoon, N.; Zhang, Z.F.; Zhou, C.H.; Chu, M.Q. Macrophage membrane coated nanoparticles: A biomimetic approach for enhanced and targeted delivery. Biomater. Sci. 2022, 10, 1193–1208.
- Zeng, Y.P.; Li, S.F.; Zhang, S.F.; Wang, L.; Yuan, H.; Hu, F.Q. Cell membrane coated-nanoparticles for cancer immunotherapy. Acta Pharm. Sin. B 2022, 12, 3233–3254.
- Xu, C.; Liu, W.; Hu, Y.; Li, W.P.; Di, W. Bioinspired tumor-homing nanoplatform for co-delivery of paclitaxel and siRNA-E7 to HPV-related cervical malignancies for synergistic therapy. Theranostics 2020, 10, 3325–3339.
- Cai, J.X.; Liu, J.H.; Wu, J.Y.; Li, Y.J.; Qiu, X.H.; Xu, W.J.; Xu, P.; Xiang, D.X. Hybrid Cell Membrane-Functionalized Biomimetic Nanoparticles for Targeted Therapy of Osteosarcoma. Int. J. Nanomed. 2022, 17, 837–854.
- Suski, J.M.; Lebiedzinska, M.; Wojtala, A.; Duszynski, J.; Giorgi, C.; Pinton, P.; Wieckowski, M.R. Isolation of plasma membrane-associated membranes from rat liver. Nat. Protoc. 2014, 9, 312–322.
This entry is offline, you can click here to edit this entry!