Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Type 2 diabetes mellitus (T2DM) is a metabolic disorder that impairs insulin signaling. Mitochondrial dysfunction causes various diseases, including cardiovascular diseases, metabolic disorders, and cancer. Mitochondria are dynamic and adjust their functions to match cellular conditions through processes collectively known as mitochondrial dynamics, which determine mitochondrial health and vitality. Dysregulated mitochondrial dynamics play essential roles in the pathophysiology of insulin resistance, obesity, and T2DM, including imbalanced dynamics in T2DM.
- fission
- fusion
- metabolic disorders
- mitochondrial biogenesis
- mitochondrial dynamics
- type 2 diabetes mellitus
1. Introduction
Diabetes mellitus, known as “diabetes”, is a critical challenge for public health and a therapeutic conundrum. Diabetes comprises a collection of metabolic conditions characterized and identified by hyperglycemia due to the deficiency of insulin secretion, impaired insulin action, or both [1]. There are two primary forms of diabetes: type 1 diabetes (T1DM), defined by insulin insufficiency, and immune-mediated pancreatic β-cell destruction. It manifests during the onset of childhood and in the formative phase of adulthood. The most prevalent form of diabetes is type 2 diabetes (T2DM) [2][3]. It is associated with insulin resistance in the early stage, followed by various degrees of β-cell dysfunction, and frequently co-occurs with other metabolic diseases, such as obesity. Diabetes and obesity are two complex multi-factorial, progressive disorders, with obesity the leading independent risk factor for developing T2DM, accounting for more than 90% of diagnoses [2][3]. Before T2DM was clinically diagnosed, there was a significant pancreatic β-cells loss in response to the loss of insulin sensitivity in peripheral tissue, primarily skeletal muscle, adipose tissue, and liver. The remaining β-cells cannot sustain secreting insulin to compensate for insulin resistance at a certain point. The overt T2DM manifests with hyperglycemia and the deterioration of the β-cell mass.
The “powerhouse of the cell,” mitochondria are cellular organelles. The word “mitochondria” comes from two Greek words that denote thread “mitos” and granule “chondros”. Most eukaryotic cells contain highly active mitochondria. It is hypothesized that mitochondria are the descendants of a prokaryote formed in a prehistoric era that underwent an endosymbiotic relationship with early eukaryotes [4]. Translocase of the outer mitochondrial membrane complex, sorting and assembly machinery complex, and porins are three essential protein families in the outer membrane. The oxidative phosphorylation enzyme complexes are located in cristae, formed by folding the inner membrane. The tricarboxylic acid (TCA) cycle is a process that happens in the mitochondrial matrix. The 16.5 kb double-stranded closed circular DNA mitochondrial genome is also found in the matrix. The mitochondrial genome comprises two ribosomal RNAs (12S and 16S rRNA), 13 OXPHOS proteins, 22 transfer RNAs, and 37 genes. Approximately 1500 mitochondrial proteins are encoded by nuclear genes and transported into the mitochondria from the cytoplasm [5].
The dominant role of mitochondria is energy conversion. To sustain bioenergetics and cell energy metabolism, adenosine triphosphate (ATP), which is synthesized by aerobic respiration in the mitochondrion, is required. TCA cycle and oxidative phosphorylation (OXPHOS) are the main series of events involved in the generation of ATP. These molecular mechanisms regulate biological system functions that necessitate the consumption and production of energy, such as integrating multiple metabolic pathways, regulating cellular apoptosis, and perpetuating the calcium homeostasis mechanism [6]. Similarly, mitochondria execute a multitude of roles that contribute to cellular metabolism. Metabolic precursors for macromolecules such as lipids, proteins, DNA, and RNA are produced by mitochondria. Reactive oxygen species (ROS) and ammonia are metabolic byproducts of mitochondria. They also can eliminate or utilize these biological byproducts. The modulation of innate immunity, the regulation of stem cells, the administration of programmed cell death, and aging are all processes in which mitochondria play a significant role. Hence, any potential mitochondrial disruption affects energy homeostasis and regulates cellular metabolism.
The primary dynamic activities include biogenesis (generating two identical healthy mitochondria from a pre-existing one after full growth and completed mitochondrial DNA replication), transportation (directed movement inside a cell), fission (segregation of a single organelle into two heterogenous mitochondria with mtDNA replication), fusion (the combining of two organelles into one), and mitophagy (targeted elimination through the autophagic pathway) [7][8][9][10][11]. Each dynamic process is essential for preserving a robust mitochondrial population, demonstrated to be crucial in both healthy physiology and disease states in organisms ranging from single-celled eukaryotes such as yeast to mammalians [12][13]. Hence, mitochondrial dysfunction can induce the pathogenesis of a wide range of seemingly unrelated disorders [14][15].
“Mitochondrial dysfunction” is caused by a metabolic imbalance of nutrient signal intake, energy production, and/or oxidative respiration [16], which in turn causes mitochondria-associated metabolic disorders. The traditional definition of mitochondrial malfunction is their inability to produce and maintain adequate quantities of ATP [17]. The term is also used to describe the unfavorable physiological reactions of mitochondria to metabolic disturbances, including irregularities in substrate catabolism, calcium buffering, iron transport, mutations in mitochondrial DNA or nuclear mitochondrial genes, changes in mitochondrial dynamics, changes in size and morphology, ROS production, and apoptosis [16][18]. However, the metabolic implications depend heavily on mitochondrial fission and fusion kinetics. Therefore, this review concentrates on the complex interplays between imbalanced mitochondrial dynamics and metabolism in T2DM.
2. Mitochondrial Dynamics: Biogenesis, Transport, Fusion, Fission, and Mitophagy
The ultrastructure of the mitochondrion reflects its biological, cellular, and molecular functions. Mitochondrial dynamics, which regulate the functional equilibrium of the mitochondria, is essential for mitochondrial functioning, energetics, mobility, and its host cell’s fate. Mitochondrial dynamics vary across cell types and tissues, but the critical protein machinery that drives the process has been remarkably preserved through evolution [19]. Reduction and escalation of any of these dynamic processes due to pathological or physiological stressors results in an imbalance affecting mitochondrial function, ultimately leading to multiple disorders in cardiovascular, neurodegenerative, metabolic diseases, and cancer [20][21].
2.1. Mitochondrial Biogenesis
Mitochondrial biogenesis (MB) occurs in healthy eukaryotic cells to generate new mitochondria and increase the number of mitochondria via dividing the pre-existing ones, which already fully grow and their DNA replication is fully completed [22]. This process involves the expression of more than one thousand genes from both mitochondrial and nuclear genomes [23] and coordinates with cell cycle events but is not limited to cell division only [24]. Peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1-α (PGC-1α) plays as the master regulator of MB. As shown in Figure 1, after activated via either phosphorylation or deacetylation by various signaling pathways such as receptor tyrosine kinases, natriuretic peptide receptors, and nitric oxide through cyclic guanosine monophosphate (cGMP), and cyclic adenosine monophosphate (cAMP), PPAR, Akt, sirtuin 1 (SIRT1)-mediated deacetylation, AMP/ATP via cyclic adenosine monophosphate (cAMP), AMP-activated protein kinase (AMPK), calcineurin/calmodulin, and specificity protein 1 (Sp1) [25][26], PGC-1α initiates MB pathway by sequential activations of nuclear transcription factors including nuclear respiratory factors 1, 2 (NRF1, NRF2), and estrogen-related receptor-α (ERR-α), followed by the increase in the expression mitochondrial transcription factor A (TFAM), then mitochondrial DNA (mtDNA) replication and transcription [27][28]. Translating mtDNA-encoded genes requires specific translation factors encoded by nuclear DNA, such as the initiation factors 2 and 3 (mtIF2 and mtIF3), elongation factors Tu, Ts, G1 (mtEFTu, mtEFTs, mtEFG1). NRF1 and NRF2, together with Sp1, bind to the gene of mitochondrial transcription factor A (TFAM) and promote the expression of TFAM, which is then imported via the translocase of the outer and inner mitochondrial membrane into mitochondria, where it regulates 13 mitochondrial genes encoding for essential components of ETC [26][29][30]. The mitochondrial proteins encoded by nuclear DNA are transported into the mitochondria via the translocase of the outer mitochondrial membrane/the translocase of the inner complex (TOM/TIM) (Figure 1). The regulation of MB is not limited to controlling PGC-1α activation through post-translational modifications but also PGC-1α transcription. Various signalings, including Akt, p38MAPK, Calcineurin A (CnA), calmodulin-dependent protein kinase IV (CamKIV), and protein kinase A (PKA) [31], as illustrated in Figure 1.
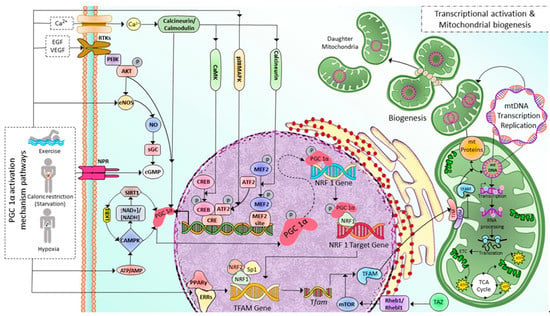
Figure 1. Illustration depicting implicated PGC 1α activation mechanism pathways and associated transcriptional modifications in regulating mitochondrial biogenesis in skeletal muscle cells. Exercise and caloric restriction promote Ca2+ entry, tyrosine kinase receptor activation, eNOS production, NPR activation, and decreased ATP/AMP ratio. All these effects, in turn, activate PGC-1α in several mechanisms either by post-translational modifications (deacetylation, phosphorylation) or by regulating PGC-1α transcription. First, PGC-1α increases NRF1 expression, then PGC-1α and NRF1 bind to the target gene and initiate the transcription of the TFAM gene. Exercise also increases the PPARγ-ERRs complexes, which enhance TFAM gene transcription. Tfam mRNA is then translated into TFAM protein; this process is facilitated by mTOR, a downstream target of TAZ-Rheb1/Rhebl1 signaling. Finally, TFAM is transported into mitochondria through TOM/TIM complexes, where it induces mitochondrial DNA replication, transcription, and translation leading to mitochondrial biogenesis. Ca2+: calcium ion; EGF: Epidermal growth factor, VEGF: Vascular Endothelial Growth Factor, RTKs: Tyrosine kninase receptors, NPR: Natriuretic peptide receptor, PI3K: Phosphoinositide 3-kinase, NO: Nitric Oxide, eNOS: edothelial Nitric Oxide, sGC: soluble Guanylyl cyclase, cGMP: cyclic guanylyl mono phosphate, SIRT1: Sirtuin 1, LKB1: Liver Kinase B1, PGC-1α: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha, NAD+/NADH: Nicotinamide Adenine Dinucleotide/Nicotinamide Adenine Dinucleotide Phosphate, AMPK: Adenosine Monophosphate-activated Protein Kinase, ATP/ADP: Adenosine Triphosphate/Adenosine Diphosphate, CaMK: Calcium/calmodulin-dependent protein kinase, p38MAPK: p38 mitogen-activated protein kinases, CREB: cAMP-responsive element binding protein, ATF2: Activating transcription factor 2, CRE: cyclic AMP Response Element, MFF2: Myocyte Enhancer Factor-2, NRF1: Nuclear Respiratory Factor 1, Sp1: specificity protein 1, PPAR-γ/EERs: Peroxisome Proliferator Activated Receptor Gamma/Estrogen-Related Receptors, TFAM: Mitochondrial Transcription Factor A, TAZ: transcriptional coactivator with PDZ-binding motif, Rheb1/Rhebl1: Ras Homolog Enriched in Brain 1/Ras Homolog Enriched in Brain-like 1, mTOR: mammalian target of rapamycin, TOM/TIM: Translocons of the Outer/Inner membrane, mt: mitochondrial.
Among the PGC-1α activators, AMPK and SIRT1, the two metabolic sensors of the cells, are the two major pathways regulating mitochondrial biogenesis [32]. AMPK is activated as a response to different conditions that deplete cellular energy, including endurance exercise, hypoxia, starvation (primarily glucose), and mitochondrial electron transport chain (ETC) inhibitors [33][34][35][36][37][38]. AMPK either directly phosphorylates PGC-1α [39][40] or activates SIRT1 (an NAD+-dependent histone/protein deacetylase) through nicotinamide phosphoribosyl transferase which in turn increases NAD+ levels [41][42]. In contrast, SIRT1 also plays a role in AMPK activation [43]. SIRT1 activates AMPK through the deacetylation of Liver Kinase B1 (LKB1), the master upstream kinase, directly phosphorylating and activating AMPK and various related kinases [44][45]. A high NAD+/NADH ratio leads to the phosphorylation of SIRT1 at thr-522 residue. Phosphorylated SIRT1 activates PGC-1α by removing the acetyl group on it. Furthermore, the MB is initiated in response to cellular stress, such as oxidative stimulus, to an increase in the energy requirements of the cells [46] and external stimuli, including nutrients, hormones, and exercise [47][48][49]. As a result, MB increases the number of healthy mitochondria, resulting in a greater metabolic capacity to match the cell’s energy needs.
A recent study discovered a novel stimulator of MB called a transcriptional coactivator with PDZ-binding motif (TAZ) [50]. PGC-1α and TAZ act at different levels in TFAM production. PGC-1α induces Tfam gene transcription. Meanwhile, TAZ promotes the translation of Tfam mRNA via Ras homolog enriched in brain (Rheb)/Rheb-like-1 (Rhebl1)- Mammalian target of rapamycin complex 1 (mTORC1) axis. TAZ also plays a vital role in the translational regulation of other mitochondrial genes [27][50][51]. Moreover, in skeletal myocytes derived from muscle-specific TAZ-knockout (mKO) mice, the Tfam protein level is decreased and not induced after exercise. However, there is no change in PGC1α, NRF1, or NRF2 levels [50].
2.2. Mitochondrial Transport
Mitochondrial transport through the cytoskeleton is crucial for normal mitochondrial morphology, network, motility, and distribution [52]. The cytoskeletal elements, actin microfilaments, and microtubules are necessary to transport and distribute mitochondria. The motor-based mitochondrial movement allows the mitochondria to navigate along microfilaments and tubules. Mitochondria bind to specific motor isoforms through adaptors explicitly designed for each organelle. In order to act as receptors and localize the motor adaptor complex to the organelle, adaptors probably need integrated outer mitochondrial membrane proteins. Thus, adaptor proteins that combine with the motors set up organelle-specific connections. Alteration of these motor and adapter proteins can impair the mitochondrial transport process. In multicellular eukaryotes, mitochondria use microtubule motors, such as the kinesin/dynein motor (Figure 2), which consists of plus-end-directed kinesins and minus-end-directed dyneins [53][54].
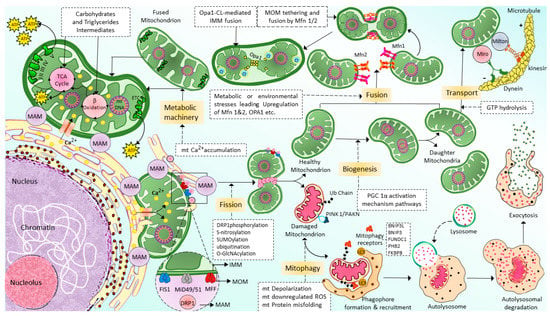
Figure 2. Schematic of the machinery and context of mitochondrial dynamics: Fission, fusion, transport, and mitophagy. Mitochondria generated from mitochondrial biogenesis are transported and distributed through the cells via cytoskeletal elements, including actin microfilaments and microtubules by kinesin and dynein motors. Mitochondrial fusion is a highly regulated and complex process in which two mitochondria are fused into one mitochondrion to maximize the OXPHOS capacity of the cells under conditions of high energy demand. Mfn1/2 is responsible for the fusion of MOMs; meanwhile, Opa1 is responsible for the fusion of IMMs. In contrast to fusion, mitochondrial fission allows a mitochondrion to segregate into two heterogenous mitochondria and enable autophagy removal of the damaged mitochondrial. Drp-1 is the primary protein that is responsible for mitochondrial fission. However, additional proteins might also be necessary for this process, such as FIS1, MFF, and MiD49/51. Finally, the damaged, defective mitochondria generated by mitochondria fission are eliminated via a selective autophagy process known as mitophagy. (For more details, please see the main text). TCA: tricarboxylic acid, MAM: mitochondria-associated ER membrane, Ca2+: calcium, mt: mitochondrial, ETC: electron transport chain, ATP: adenosine triphosphate, GTP: guanylyl triphosphate, Opa1: optic atrophy 1, Mfn1/2: mitofusin 1/2, Drp1: dynamin-related protein 1, FIS1: mitochondrial fission 1 protein, MFF: mitochondrial fission factor, and MiD49/51: mitochondrial dynamin protein 49/51, MOM: mitochondrial outer membrane, IMM: inner mitochondrial membrane, ROS: reactive oxygen species, ub: ubiquitination, PINK1/PAKN: PTEN-induced putative kinase protein 1/Parkin-mediated ubiquitination, BNIP3L: BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3, FUNDC1: FUN14 domain containing 1, PHB2: Prohibitin-2, FKBP8: FK506-binding protein 8.
2.3. Mitochondrial Fusion
Mitochondrial fusion is a complex regulatory mechanism involving several proteins’ binding from multiple mitochondrion membranes (Figure 2). When cells experience metabolic or environmental stress, mitochondria undergo fusion by cointegrating partially damaged mitochondria, lowering the stress by optimizing ATP production [55]. The outer membrane (MOM) of the mitochondria contains mitochondrial fusion factor-mitofusins, a member of the hydrolase enzyme family. Mitofusins are essential for the fusion of the MOM. The ocular atrophy protein 1 (OPA1), anchored to the inner mitochondrial membrane (IMM), allows the IMM to undergo fusion. The cristae organization is necessary to produce cellular energy, the mitochondrial ETC, mitochondrial membrane potential (ΔΨm) generated by proton pumps, regulation of cell apoptosis, and maintenance of mtDNA are modulated by OPA1 [56]. MFN1, MFN2, and OPA1 join the inner and outer membranes of mitochondria during the fusion mechanism. In certain cellular scenarios, a functional substitution between MFN1 and MFN2 occurs. This is brought on by cells lacking MFN1 inducing MFN2 to overexpress and vice versa [57]. There is a significant relationship between cellular physiological activity and mitochondrial fusion. During the fusion mechanism, the old mitochondrial transfers proteins and mtDNA to newly formed mitochondria, which in turn assist in limiting the accumulation of damaged mtDNA [58]. Due to ROS-induced mutations in mtDNA [59], some mitochondria are unable to perform their respiratory activity optimally in order to improve the fusing mitochondria’s overall respiratory function. Therefore, mitochondrial fusion is essential because it enables the interchange of gene products and metabolites. Similarly, diminished mitochondrial function is linked to a restriction of mitochondrial fusion [60]. Inhibition of fusion impairs OXPHOS, induces mtDNA depletion, and elevates ROS production [61]. The disruption of the fission mechanism causes imbalanced fusion, leading to an increase in the number of elongated mitochondria. However, interference with the fusion process leads to more fragmented mitochondria [62].
2.4. Mitochondrial Fission
Mitochondrial fission segregates a mitochondrion into two heterogenous mitochondria without mDNA (Mitochondrial DNA) replication (Figure 2). Fission is crucial for removing damaged organelles by mitophagy. Mitochondrial division, size, and form, as well as the distribution of mitochondria throughout the cell, depending on mitochondrial fission. There are two major ways that mitochondria undergo fission: Mitochondrial Fission Process 1 (MTFP1), in which the IMM splits first and eventually causes the separation of OMM, and Mitochondrial Fission Process 2 (MTFP2), in which the mitochondria accumulate from the fission point inward and separate. Dynamin-related protein 1 (DRP1), a GTPase fission mediator, is recruited from the cytosol and proceeds to the MOM, forming foci and rings surrounding mitochondria to mediate the fission process. Currently, most of the proteins which are known to be associated with mitochondrial fission are GTPase family proteins, including DRP1 itself and others such as mitochondrial fission 1 protein (FIS1), mitochondrial fission factor (MFF), and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51). MiD49 and MiD51 are anchored in the MOM surface [63][64][65][66]. The structure of the DRP1 includes an N-terminal GTPase domain followed by the middle domain, variable domain (or Binsert), and the GED in the C-terminal. A critical mechanistic process of systolic Drp1 recruitment into the MOM surface is due to the proposed act as receptors in the MOM. The MOM receptors in this process are FIS1, MFF, MiD49, and MiD51 [67][68].
2.5. Mitochondrial Selective Autophagy (Mitophagy)
Mitophagy [69], also known as mitochondrial selective autophagy [70], is crucial for reestablishing cellular balance in both normal physiology and during stressful circumstances [71]. Mitophagy is a process that eliminates the damaged or extra mitochondria as double-membraned autophagosomes for subsequent lysosomal degradation. Mitochondrial fragmentation by fission is one of the crucial processes associated with mitophagy [72][73]. Hence, mitophagy and mitochondrial fission regulate the abundance of mitochondria and their functionality in the host cells [74]. Mitophagy is a multi-stage process that involves numerous phases (Figure 2). They can be categorized as follows: mitophagy initiation phase, mitochondrial labeling for autophagy machinery detection, production of autophagosomes for labeled mitochondria engulfment, lysosomal trapping, and enzyme hydrolysis [75]. Multiple regulatory mitophagy mechanisms are currently induced in response to various stimuli. They are typically divided into two categories: receptor-mediated mitophagy and PTEN-induced putative kinase protein 1 (PINK1)/Parkin-mediated ubiquitination (PAKN) mitophagy pathway [76]. In response to various mitochondrial stressors, receptor-mediated mitophagy is controlled at the transcriptional or post-transcriptional level [70]. The mitophagy receptors residing in the MOM can attract autophagosomes to mitochondria. This is the main characteristic feature of mitophagy receptors [77][78]. BCL2 Interacting Protein 3-like (BNIP3L or NIX), BCL2 interacting protein 3 (BNIP3), FUN14 domain containing 1 (FUNDC1), Prohibitin2 (PHB2), and FKBP Prolyl Isomerase 8 (FKBP8) are the few examples of mitophagy. At least one LC3 interacting region (LIR), which can directly bind to the autophagy mediator LC3 and attract autophagosomes to mitochondria, is a characteristic of mitophagy receptors [79].
The most well-known mitophagy pathway that controls mitochondrial maintenance and quality control is the PINK1/PAKN mitophagy pathway [71]. PINK1 is a serine/threonine kinase with a mitochondrial target sequence (MTS) and transmembrane domain (TMD), and PAKN is a cytosol ubiquitin E3 ligase [74]. Together, PINK1/PAKN detects cellular stress and coordinates the elimination of damaged mitochondria. PINK1 is continuously imported into mitochondria under physiological circumstances with normal mitochondrial membrane potential, where it is cleaved by the intramembrane protease presenilin-associated rhomboid-like (PARL) protein, causing its retro-translocation into the cytosol and quick proteasomal degradation [80][81].
3. Mitochondrial Dynamics in T2DM
T2DM and its vascular complications are associated with mitochondrial dynamics. Mitochondria acts as an important regulator of insulin secretion. In addition, Mitochondria are the primary generators of ROS and play a central role in cellular apoptosis and cell death. High glucose levels in T2DM increase ROS production [82][83][84], promoting oxidative stress and activating various signalings mediating the micro and macrovascular complications in diabetes [83][85][86]. High-calorie intake causes intramyocellular lipid accumulation [87][88][89], enhancing ROS production, oxidative stress, and mitochondrial dysfunction in the skeletal muscle [90][91][92]. Obesity disrupts the β-oxidation of FA into acetyl-CoA to supply the substrates for utilization in the TCA [93]. Decreased β-oxidation causes cellular dysfunction due to the synthesis of triglycerol and ectopic deposition of lipids. Increased free FA also promotes ROS production resulting from the increase in lipid peroxidation byproducts [91][94], the effect known as lipotoxicity. The activities of antioxidant enzymes such as glutathione S-transferase, glutathione peroxidase, paraoxonase-1, and catalase decrease, which, therefore, superimpose the oxidative stress condition in different tissues of obese rats [95]. Therefore, ROS is an important mediator in dysregulated mitochondrial function and T2DM. The changes in mitochondrial morphology and dynamics are required to increase ROS under hyperglycemia [96]. Therefore, mitochondrial dynamics are a major regulator of mitochondrial function. Skeletal muscle derived from obesity and T2D individuals have an impaired functional capacity of mitochondria correlated with decreased mitochondrial size, number, and damaged cristae [96][97]. Hyperglycemia also induces the disruption of mitochondria in other cell types, including the heart, vascular, and liver [96][98][99]. However, glucose metabolic disorders do not always play the causal factor; it might also be the dysregulated mitochondrial dynamics [100][101][102], as discussed later.
Insulin signaling promotes the expression of Opa-1 protein via the Akt-mTOR-NFkB-Opa-1 pathway, leading to mitochondrial fusion [103]. In T2DM, this signaling is disrupted. Therefore Opa-1 expression level decreases, leading to imbalanced fission, reducing ATP production efficiency, and causing cellular dysfunction. Reduced MFN2 expression is associated with T2DM and may be connected to skeletal muscle mitochondrial dysfunction. Furthermore, it has recently been shown that in leukocytes from T2D patients, mitochondrial fusion was reduced, and fission was enhanced, especially in patients with poor glycemic control [104]. In addition, the activation of leukocyte–endothelial contacts in diabetes patients was associated with lower mitochondrial fusion and higher mitochondrial fission. Leukocyte rolling flux also increased concurrently with HbA1c levels [104]. These findings imply that glycemic management affects mitochondrial dynamics in the leukocytes of diabetic patients, where there is an increase in leukocyte–endothelial contacts and a decrease in mitochondrial fusion (Figure 3). Therefore, it is theorized that having T2D with poor glycemic management changes mitochondrial dynamics, encouraging leukocyte–endothelial interactions and the development of cardiovascular diseases and other vascular complications [103][104]. Moreover, hyperglycemia impaired endothelial function directly by promoting mitochondrial fission, iNOS activation, and ROS production [105].
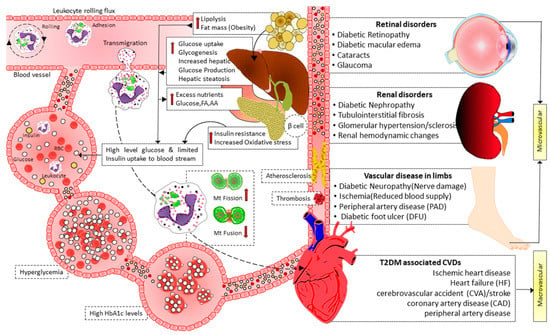
Figure 3. Illustration showing the interplay of mitochondrial dynamics in activating leukocyte–endothelial contacts and micro and macrovascular complications of diabetes. Hyperglycemia in T2D induces mitochondrial fission and decreases mitochondrial fusion, facilitating leukocyte activation and endothelial interactions. Furthermore, high blood glucose levels also cause mitochondrial fission, impairing endothelial function via activating iNOS and increasing ROS production. As a result of leukocyte activation, leukocyte–endothelial interactions, endothelial dysfunction, atherosclerosis, and thrombosis mediate micro- and macro-vascular complications in T2D. FA: fatty acid, AA: amino acid, Mt: mitochondrial, HbA1c: hemoglobin A1c, T2D: type 2 diabetes, CVDs: cardiovascular diseases.
White adipose tissue derived from ob/ob mice (obese mice model) exhibit a fusion-to-fission balance reflected by the increase in Drp-1 and the decrease in MFN2 and OPA1 protein expression, decreased mitochondrial biogenesis evidenced by a reduced content of mitochondrial DNA and PGC-1α mRNA expression. Treatment with either leptin or mitochondrial division inhibitor (mdivi-1) improved blood glucose levels, lipid oxidation, and mitochondrial function by storing mitochondrial dynamics balance [106].
Moreover, T2D dyslipidemia models exhibit elevated mitochondrial fission due to mitochondrial depolarization, decreased ATP synthesis, elevated oxidative stress, and decreased insulin-stimulated glucose uptake. Insulin resistance and mitochondrial fragmentation brought on by excessive palmitate were reduced by both genetic and pharmaceutical suppression of DRP1 [107]. In another study, DRP1 was elevated in rat pancreatic islets after stimulation by FFAs, and this DRP-1 overexpression was accompanied by increased β-cell death [108]. Many processes involved in atherosclerosis (a common complication of T2D) are linked to mitochondrial fission, including endothelial dysfunction [105][109][110][111][112][113], collagen matrix [114][115], and the motility and proliferation of vascular smooth muscle cells [116][117][118][119]. In endothelial cells derived from T2D patients, silencing FIS1 or DRP1 inhibited high glucose-induced mitochondrial fission and ROS generation [105]. Metformin prevents the DRP1-mediated mitochondrial fission that leads to atherosclerosis in diabetic mice via an AMP-activated protein kinase (AMPK)-dependent pathway [120]. DRP1 is essential in the pathogenesis of diabetic micro and macrovascular complications [121][122][123][124][125][126][127][128][129][130][131][132][133][134][135].
Metabolic disorders affect mitochondrial fusion/fission balance and impair biogenesis [136][137][138][139]. Downregulation of PGC-1α (the master regulator of metabolism and mitochondrial biogenesis) leads to mitochondrial damage and decreased mitochondrial density in obesity. The serum concentrations of adiponectin were reduced in obese subjects. The adiponectin can stimulate deacetylation and transcriptional activity of PGC-1α and MB via engaging to adiponectin receptor to activate Ca2+ and AMPK/SIRT1 signaling [140]. Therefore, the downregulation of PGC-1α gene expression or activity is implicated in obesity and diabetes [141][142][143]. Physical exercise can improve insulin resistance by restoring PGC-1α activation and expression; hence, MB is mediated by increased intracellular Ca2+ and AMPK/SIRT1 signaling (Figure 1).
4. Dysregulated Mitochondrial Dynamics and Metabolism Play a Causal Role in Diabetes
Mitochondrial dysfunction due to imbalanced mitochondrial dynamics leads to decreased OXPHOS, ATP production, β-oxidation of FFA [97][144], and enhanced ROS production [145]. Mitochondrial dysfunction plays a causal role by governing insulin secretion failure in β-cells and peripheral insulin resistance through the gluco-lipotoxicity [5][6] (Figure 4) or by impairing the regulation of glucose homeostasis in neurons of different cerebral regions [92][146][147][148][149][150] and glial cells in the brain (Figure 5). Excessive energy supply and low ATP demand increase mitochondrial fission, proton leak, and redox imbalance. On the other hand, in the depletion of nutrient supply and high ATP demand, the mitochondria are hyperfused, and respiration is optimized [151]. Accumulation of lipids/FFA increases diacylglycerol (DAG) and ceramides which activate protein kinase C (PKC) and protein phosphatase 2 (PP2A), respectively. PKC phosphorylates and inhibits insulin receptors. Meanwhile, PP2A dephosphorylates Akt and impairs insulin’s downstream signaling in which glucose transporter translocates to the cell membrane [152][153][154]. Furthermore, increased oxidative stress induces mitophagy and apoptosis, exacerbating the decrease in β-oxidation of FFA further, and ROS promotes Serine/threonine-specific protein kinases (Ser/Thr kinases) which phosphorylate and inhibit insulin receptor substrate (IRS) [155][156]. All these effects finally cause insulin resistance [157].
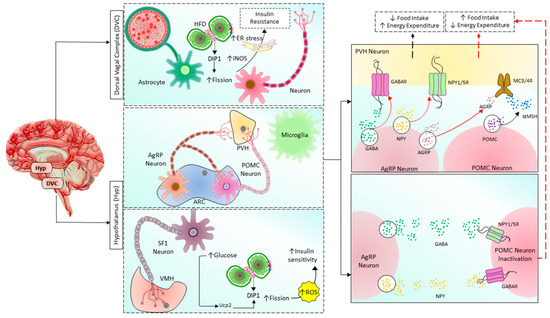
Figure 4. Mitochondrial fusion/fission balance plays a vital role in regulating the body’s metabolism by the brain in the hypothalamus and DVC. Hypothalamus has two nuclei involving energy homeostasis: ARC and VMH. By controlling PVH neurons, POMC and AgRP neurons regulate food intake and energy expenditure. POMC neurons release POMC, which is then cleaved into α-MSH and stimulates VMH neurons via MC3/4R, finally promoting negative energy balance by inhibiting food intake and increasing energy expenditure. In contrast, AgRP neurons directly inhibit VMH neurons via releasing AgRP, GABA, and NPY, acting on MC3/4R, GABAR, and NPY1/5R, respectively, finally promoting positive energy balance by increasing food intake and reducing energy expenditure. AgRP neurons also indirectly promote positive energy balance by inhibiting POMC neurons through GABAR and NPY1/5R. In addition, in SF1 neurons of VMH, increased glucose level induces mitochondrial fission followed by ROS production and insulin resistance in a upc2-dependent manner. On the other hand, excessive nutrients in the astrocytes and neurons located in DVC cause insulin resistance by increasing mitochondrial fission, iNOS activity, and ER stress. HFD: high-fat diet, ER: endoplasmic reticulum, Drp1: dynamin-related protein 1, iNOS: inducible nitric oxide synthase, AgRP: Agouti-related peptide, POMC: Pro-opiomelanocortin, PVH: paraventricular nucleus of the hypothalamus, ARC: arcuate nucleus, SF1: steroidogenic factor-1, ROS: reactive oxygen species, GABA: Gamma-aminobutyric acid, GABAR: GABA receptor, NPY: neuropeptide Y, NPY1/5R: NPY receptor 1/5, α-MSH: α-melanocyte-stimulating hormone, MC3/4R: melanocortin-3/4 receptor.
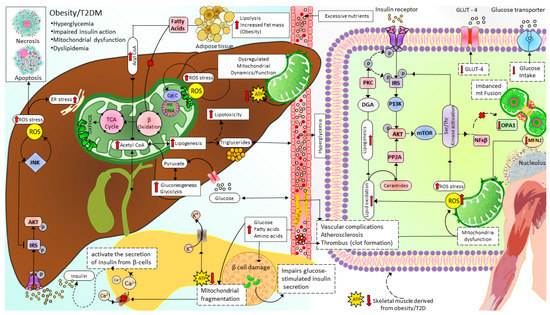
Figure 5. Mitochondrial dynamics and metabolism in the pathophysiology of T2D. Excessive nutrients (for example, HDF) induce imbalanced mitochondrial dynamics leading to decreased OXPHOS capacity and accumulation of FA and lipotoxicity in which ceramides and DAG impair insulin signaling. In addition, reduced ATP/ADP ratio in pancreatic β-cells disrupts glucose-stimulated insulin secretion, further increasing blood glucose levels. T2D: type 2 diabetes, ROS: Reactive oxygen species, IRS: Insulin receptor substrate signaling, AKT: serine/threonine kinase pathway, JNK: c-junN-terminal kinase pathway, ER: endoplasmic reticulum, TCA cycle: tricarboxylic acid cycle, ATP: adenosine triphosphate, Ca2+:calcium ion, β oxidation: beta-oxidation of fatty acid (Fatty acid cycle), GLUT4: glucose transporter type 4, P13K: phosphatidylinositol-3 kinase, PP2A: protein phosphatase 2A, PKC: protein kinase C, DGA: diacylglycerol, mTOR: mammalian target of rapamycin, NFKβ: nuclear factor kappa light chain enhancer of activated beta cells, OPA1: optic atrophy-1, MFN2: mitofusin2.
Chronic exposure to high levels of glucose, FA, and amino acids damages β-cells. It impairs glucose-stimulated insulin secretion due to mitochondrial fragmentation, which decreases the production of ATP (increased ATP/ADP ratio close ATP-sensitive K+ channels and depolarization of membrane potential initiates, followed by increased calcium levels which activate the secretion of insulin from β-cells) [158].
The previous study in cybrid b4 discovered that overexpression of MFN1/2 or DRP1/FIS1 knockdown increased mitochondrial fusion, insulin signaling, and Glu4 translocation to membranes [159]. In contrast, MFN1/2 knockdown or DRP1/FIS1 overexpression reduced the mitochondrial network (promoted fission) and decreased IRS-AKT signaling and Glut4 translocation [159]. Furthermore, a later study reported that excessive nutrients also increased chronic inflammatory markers, inflammasome, and these effects were abolished by overexpression of fusion proteins (Mfn1 or Mfn2) [160]. In another study on L6 rat-derived skeletal muscle cells, silencing MFN2 and OPA1 induced mitochondrial fragmentation, attenuation of cellular respiration, and insulin-dependent Akt phosphorylation [161]. The results from this study suggest that the imbalance of mitochondrial fusion and fission plays a causal role in the pathogenesis of insulin resistance and T2DM.
In mammals, the hypothalamus and dorsal vagal complex (DVC) have been implicated as crucial regulators of whole-body metabolism [149][150][162]. In the hypothalamic arcuate nucleus, there are populations of neurons known as arcuate melanocortin neurons that regulate the body’s metabolism. The first group secretes agouti-related peptide (Agrp) and neuropeptide-Y (NPY), which promote food intake and decrease energy expenditure [163][164][165][166][167]. These neurons’ activity increases when the body’s energy is depleted [168][169][170][171][172]. The second group is the pro-opiomelanocortin (POMC)-expressing neurons that function oppositely in regulating metabolism. The difference in functions of these two neuronal populations is due to the opposite effects on their target neurons, the melanocortin 3/4 receptor (MC3/4R)-expressing neurons in the paraventricular nucleus of the hypothalamus (PVH) (Figure 4). POMC neurons release α-melanocyte stimulating hormone (α-SMH, a cleaved product of POMC), which binds to and activates MC4R in the paraventricular nucleus of the hypothalamus to inhibit food intake and promote energy expenditure [173]. AgRP neurons release AgRP, which inhibits MC3/4R signaling, stimulating food intake and reducing energy expenditure [164][166][167][174]. Interestingly, these neurons also release NPY and neurotransmitter gamma-aminobutyric acid (GABA), which inhibit POMC neurons via neuropeptide Y receptor 1 and 5 (NPY1R and NPY5R) and GABAA receptors, respectively [175][176][177][178][179]. Therefore, once activated, AgRP neurons can amplify their effects by inhibiting their antagonizing neurons. The energy state reciprocally regulates POMC and AgRP neurons, the adiposity hormones leptin, and insulin. AgRP neurons are inhibited by leptin and insulin [180][181][182][183] and activated by the gastric hormone ghrelin [184][185][186]. In contrast, leptin and insulin activate POMC neuron signals under high energy demand [175][187][188][189][190][191]. Neurons require high energy relative to other cells, they utilize 20% of whole-body oxygen consumption [192], and mitochondria are the primary factory that generates and supplies ATP to them. Therefore, the mitochondria have a critical role in maintaining neuronal activities. Mitochondrial functions are reflected by their morphology and dynamics, which can be changed to support cellular energy homeostasis in response to different conditions. As mentioned above, the balance of mitochondrial dynamics in neurons supports cellular function and neuronal activities to control energy homeostasis [193]. Schneeberger et al. found that POMC-specific Mfn knockout mice fed the HF diet had fewer mitochondrial-ER contacts, decreased mitochondrial length, and branching in POMC neurons [150]. Specific MFN2 knockout in these neurons decreased α-MSH release, altered mitochondrial morphology, decreased mitochondrial-ER contacts and ER stress, reduced the conversion of POMC into α-MSH, developed leptin resistance due to ER stress, increased ROS production due to impaired complex I activity, increased energy intake, decreased energy consumption, and finally lead to obesity [150]. These effects can be reversed by chemical chaperones such as 4-phenyl butyric acid (4-PBA) or tauroursodeoxycholic acid (TUDCA), which relieves ER stress [150]. These results suggested that MFN2 in POMC neurons is the key regulator of body energy homeostasis via mitochondrial-ER axis homeostasis and function. Surprisingly, POMC-specific OPA1-knockout mice develop obesity at 7 weeks of age and exhibited mitochondrial cristae loss fragmentation, Ca2+ overload, and decreased α-MSH release, but glucose sensing, neuronal activation, and hypothalamic ROS production are not changed [148][194]. DRP1-mediated mitochondrial fission negatively regulates POMC neuronal responses to glucose and leptin sensitivity contributing to obesity and diabetes development. Selective Drp1 knockout in POMC neurons inhibits mitochondrial fission and improves glucose metabolism and leptin sensitivity [195]. More detailed insights into the role of mitochondrial dynamics in the hypothalamic regulation of glucose and energy homeostasis can be found in Sungho Jin and Sabrina Diano’s review [196].
Recent studies also found that under excessive nutrients, Drp1 is activated, mediates mitochondrial fission in DVC neurons, and induces insulin resistance [92]. Inhibition of Drp1 by infusion of mitochondrial division inhibitor 1 (MDIVI-1) reverses HFD-induced mitochondrial fragmentation, endoplasmic reticulum (ER) stress, and insulin resistance in DVC neurons. Furthermore, activation of Drp1 alone is sufficient to cause mitochondrial fission, ER stress in DVC neurons, and insulin resistance. ER stress is the critical mediator in mitochondrial fission-induced insulin resistance in DVC, and relief of ER stress using 4-PBA restores insulin sensitivity in DVC of 3-day HFD rats [92]. Moreover, in a recent study, Patel et al. found that mitochondrial dynamics in DVC astrocytes also play an important role in whole-body metabolism. Increased Drp1 activity disrupts insulin signaling, increasing food intake, weight gain, and adipose tissue. Drp1 activation also increases DVC’s inducible nitric oxide synthase (iNOS) levels (Figure 4). Either inhibition of Drp1 or iNOS can prevent HFD-induced insulin resistance and obesity [197]. Astrocyte-specific conditional deletion of MFN2 suppressed perivascular mitochondrial clustering and disrupted mitochondria-endoplasmic reticulum (ER) contact sites. These results suggest that mitochondrial fission mediated by Drp1 and ER stress are essential in regulating whole-body energy homeostasis of DVC.
The activation of Drp1 and mitochondrial fission requires the functional integration between phosphorylations at S600 and at S579 sites of Drp1 [19]. S600 phosphorylation occurs first, then initiates the phosphorylation of the other one. The mice expressed S600 phosphor null form (S600A, which interferes with the phosphorylation of S600) have decreased mitochondrial fission, increased lipid oxidation and OXPHOS capacity, insulin sensitivity, and thermogenic response under HFD feeding [19]. In addition, phosphorylation at S616 promotes mitochondrial fission [198], but phosphorylation of Drp1 at S637 causes the opposite effect on mitochondrial dynamics [199][200]. Therefore, post-translational modifications of Drp1 are important in regulating mitochondrial fission. Moreover, Syntaxin 4, an exocytosis protein, enhances glucose uptake and improves insulin sensitivity in HFD diabetic mice by suppressing mitochondrial fission through phosphorylation of Drp1 at S637 in a pathway involved in AMPK [201].
In response to energy depletion due to decreased food intake or increased demand during exercise, mitochondrial biogenesis is activated to maintain and improve the number of healthy mitochondria for ATP production [50][202][203][204][205][206][207]. The disruption of mitochondrial biogenesis decreases OXPHOS and ATP production capacity and is associated with metabolic disorders, including obesity and T2DM [197][208]. PGC-1α is the central inducer of mitochondrial biogenesis [209][210] and is a potential target for modulating mitochondrial mass. The conditions of energy depletion, such as endurance exercise, activate PGC-1α activity through Ca2+/CaMK/calcineurin, AMPK/SIRT1, NO, and Akt signalings and enhance PGC-1α transcription through CaMK, calcineurin, and Akt pathways (Figure 1). In skeletal muscle during the aging of mice, Tina Wenz et al. found that PGC-1α overexpression preserved mitochondrial function, neuromuscular junction, and muscle integrity, reduced ROS production, apoptosis, autophagy, proteasome degradation, and improved insulin sensitivity in skeletal muscle of mice during aging [211]. Recent compelling evidence has shown that exercise promotes MB by activating PGC-1α [212][213][214][215] and increases mitochondrial function in human skeletal muscle [47][216][217][218][219][220]. Jun-Ha Hwang et al. recently discovered the underlying mechanism that exercises grows MB via transcriptional coactivator with PDZ-binding motif (TAZ), which induces TFAM transcription through Ras homolog enriched in brain (Rheb)/Rheb-like-1 (Rhebl1)-mTOR axis. Therefore, TAZ is responsible for MB and exercise-induced muscle adaption [50]. In alloxan, monohydrate-induced diabetic rabbits, pioglitazone (an anti-diabetic drug used to treat T2DM) improves mitochondrial biogenesis and function and reduces NF-κB and TGF-β1 expression levels via the PPAR-γ/PGC-1α pathway. These effects of pioglitazone improve diabetic atrial structural and electrophysiological remodeling. When PGC-1α is silenced using siRNA transfection, the impacts of pioglitazone are blunted [137]. These results again suggest that PGC-1α is the efficient target in regulating mitochondrial dynamics, hence mitochondrial function and metabolic homeostasis in T2DM.
ROS is involved in various pathological processes, especially in mitochondrial dysfunction and apoptosis. In the mice model, ROS activates nuclear factor-κB (NF-κB), stimulating cytokine production (tumor necrosis factor-α, interleukin 6), leading to increased lipolysis, nonesterified fatty acid, glycerol levels, gluconeogenesis, and de novo lipogenesis, finally develop insulin resistance and diabetes. In addition, NF-κB also causes p62 protein/sequestosome 1 (p62/SQSTM1) accumulation, recruiting damaged mitochondria with polyubiquitin chains and thereby inducing excessive mitophagy. These effects of ROS are ameliorated by the administration of ROS scavenger or NF-κB inhibitor .
This entry is adapted from the peer-reviewed paper 10.3390/cells12091223
References
- Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37 (Suppl. 1), S81–S90.
- Bullard, K.M.; Cowie, C.C.; Lessem, S.E.; Saydah, S.H.; Menke, A.; Geiss, L.S.; Orchard, T.J.; Rolka, D.B.; Imperatore, G. Prevalence of diagnosed diabetes in adults by diabetes type—United States, 2016. Morb. Mortal. Wkly. Rep. 2018, 67, 359.
- Xu, G.; Liu, B.; Sun, Y.; Du, Y.; Snetselaar, L.G.; Hu, F.B.; Bao, W. Prevalence of diagnosed type 1 and type 2 diabetes among US adults in 2016 and 2017: Population based study. BMJ 2018, 362, k1497.
- Archibald, J.M. Endosymbiosis and eukaryotic cell evolution. Curr. Biol. 2015, 25, R911–R921.
- Haastrup, M.O.; Vikramdeo, K.S.; Singh, S.; Singh, A.P.; Dasgupta, S. The Journey of Mitochondrial Protein Import and the Roadmap to Follow. Int. J. Mol. Sci. 2023, 24, 2479.
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta (BBA)-Bioenerg. 2017, 1858, 602–614.
- Dai, W.; Jiang, L. Dysregulated mitochondrial dynamics and metabolism in obesity, diabetes, and cancer. Front. Endocrinol. 2019, 10, 570.
- Archer, S.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251.
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287.
- Chen, H.; Chan, D.C. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 2017, 26, 39–48.
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884.
- Labbé, K.; Murley, A.; Nunnari, J. Determinants and functions of mitochondrial behavior. Annu. Rev. Cell Dev. Biol. 2014, 30, 357–391.
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646.
- Carelli, V.; Chan, D.C. Mitochondrial DNA: Impacting central and peripheral nervous systems. Neuron 2014, 84, 1126–1142.
- Lightowlers, R.N.; Taylor, R.W.; Turnbull, D.M. Mutations causing mitochondrial disease: What is new and what challenges remain? Science 2015, 349, 1494–1499.
- Kusminski, C.M.; Scherer, P.E. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol. Metab. 2012, 23, 435–443.
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312.
- Lahera, V.; de Las Heras, N.; López-Farré, A.; Manucha, W.; Ferder, L. Role of mitochondrial dysfunction in hypertension and obesity. Curr. Hypertens. Rep. 2017, 19, 11.
- Valera-Alberni, M.; Joffraud, M.; Miro-Blanch, J.; Capellades, J.; Junza, A.; Dayon, L.; Galindo, A.N.; Sanchez-Garcia, J.L.; Valsesia, A.; Cercillieux, A. Crosstalk between Drp1 phosphorylation sites during mitochondrial remodeling and their impact on metabolic adaptation. Cell Rep. 2021, 36, 109565.
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620.
- Suárez-Rivero, J.M.; Villanueva-Paz, M.; de la Cruz-Ojeda, P.; De la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; De Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Sánchez-Alcázar, J.A. Mitochondrial dynamics in mitochondrial diseases. Diseases 2016, 5, 1.
- Mullins, C. The Biogenesis of Cellular Organelles; Springer: Berlin/Heidelberg, Germany, 2005.
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 2007, 76, 701–722.
- Valero, T. Editorial (thematic issue: Mitochondrial biogenesis: Pharmacological approaches). Curr. Pharm. Des. 2014, 20, 5507–5509.
- Whitaker, R.M.; Corum, D.; Beeson, C.C.; Schnellmann, R.G. Mitochondrial biogenesis as a pharmacological target: A new approach to acute and chronic diseases. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 229–249.
- Sanchis-Gomar, F.; Luis Garcia-Gimenez, J.; Carmen Gomez-Cabrera, M.; Pallardo, F.V. Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches. Curr. Pharm. Des. 2014, 20, 5619–5633.
- Larsson, O.; Morita, M.; Topisirovic, I.; Alain, T.; Blouin, M.J.; Pollak, M.; Sonenberg, N. Distinct perturbation of the translatome by the antidiabetic drug metformin. Proc. Natl. Acad. Sci. USA 2012, 109, 8977–8982.
- Wood Dos Santos, T.; Cristina Pereira, Q.; Teixeira, L.; Gambero, A.; Villena, J.A.; Lima Ribeiro, M. Effects of Polyphenols on Thermogenesis and Mitochondrial Biogenesis. Int. J. Mol. Sci. 2018, 19, 2757.
- Marin, T.L.; Gongol, B.; Zhang, F.; Martin, M.; Johnson, D.A.; Xiao, H.; Wang, Y.; Subramaniam, S.; Chien, S.; Shyy, J.Y.-J. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci. Signal. 2017, 10, eaaf7478.
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab 2012, 23, 459–466.
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884s–890s.
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029.
- Marcinko, K.; Steinberg, G.R. The role of AMPK in controlling metabolism and mitochondrial biogenesis during exercise. Exp. Physiol. 2014, 99, 1581–1585.
- Dengler, F. Activation of AMPK under Hypoxia: Many Roads Leading to Rome. Int. J. Mol. Sci. 2020, 21, 2428.
- Chun, Y.; Kim, J. AMPK-mTOR Signaling and Cellular Adaptations in Hypoxia. Int. J. Mol. Sci. 2021, 22, 9765.
- Ren, Y.; Shen, H.M. Critical role of AMPK in redox regulation under glucose starvation. Redox Biol. 2019, 25, 101154.
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262.
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023.
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022.
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1. J. Biol. Chem. 2005, 280, 16456–16460.
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673.
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060.
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690.
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635.
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575.
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 1263–1268.
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47.
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566.
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698.
- Hwang, J.H.; Kim, K.M.; Oh, H.T.; Yoo, G.D.; Jeong, M.G.; Lee, H.; Park, J.; Jeong, K.; Kim, Y.K.; Ko, Y.G.; et al. TAZ links exercise to mitochondrial biogenesis via mitochondrial transcription factor A. Nat. Commun. 2022, 13, 653.
- Morita, M.; Gravel, S.P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711.
- Boldogh, I.R.; Pon, L.A. Interactions of mitochondria with the actin cytoskeleton. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2006, 1763, 450–462.
- Nangaku, M.; Sato-Yoshitake, R.; Okada, Y.; Noda, Y.; Takemura, R.; Yamazaki, H.; Hirokawa, N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell 1994, 79, 1209–1220.
- Tanaka, Y.; Kanai, Y.; Okada, Y.; Nonaka, S.; Takeda, S.; Harada, A.; Hirokawa, N. Targeted disruption of mouse conventional kinesin heavy chain kif5B, results in abnormal perinuclear clustering of mitochondria. Cell 1998, 93, 1147–1158.
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065.
- Griparic, L.; Van Der Wel, N.N.; Orozco, I.J.; Peters, P.J.; Van Der Bliek, A.M. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J. Biol. Chem. 2004, 279, 18792–18798.
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200.
- Chan, D.C. Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259.
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta (BBA)-Bioenerg. 2012, 1817, 1833–1838.
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192.
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383.
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99.
- Chen, K.H.; Dasgupta, A.; Lin, J.; Potus, F.; Bonnet, S.; Iremonger, J.; Fu, J.; Mewburn, J.; Wu, D.; Dunham-Snary, K.; et al. Epigenetic Dysregulation of the Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and Promotes Pulmonary Arterial Hypertension: Mechanistic and Therapeutic Implications. Circulation 2018, 138, 287–304.
- Kamerkar, S.C.; Kraus, F.; Sharpe, A.J.; Pucadyil, T.J.; Ryan, M.T. Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat. Commun. 2018, 9, 5239.
- Yu, R.; Jin, S.B.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748.
- Yu, R.; Liu, T.; Ning, C.; Tan, F.; Jin, S.-B.; Lendahl, U.; Zhao, J.; Nistér, M. The phosphorylation status of Ser-637 in dynamin-related protein 1 (Drp1) does not determine Drp1 recruitment to mitochondria. J. Biol. Chem. 2019, 294, 17262–17277.
- Civiletto, G.; Varanita, T.; Cerutti, R.; Gorletta, T.; Barbaro, S.; Marchet, S.; Lamperti, C.; Viscomi, C.; Scorrano, L.; Zeviani, M. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 2015, 21, 845–854.
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667.
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5.
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022.
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell Dev. Biol. 2020, 8, 467.
- Mao, K.; Wang, K.; Liu, X.; Klionsky, D.J. The scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev. Cell 2013, 26, 9–18.
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446.
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185.
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525.
- Gkikas, I.; Palikaras, K.; Tavernarakis, N. The role of mitophagy in innate immunity. Front. Immunol. 2018, 9, 1283.
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 2784–2790.
- Wu, H.; Chen, Q. Hypoxia activation of mitophagy and its role in disease pathogenesis. Antioxid. Redox Signal. 2015, 22, 1032–1046.
- Geng, G.; Liu, J.; Xu, C.; Pei, Y.; Chen, L.; Mu, C.; Wang, D.; Gao, J.; Li, Y.; Liang, J. Receptor-mediated mitophagy regulates EPO production and protects against renal anemia. Elife 2021, 10, e64480.
- Zuo, Z.; Jing, K.; Wu, H.; Wang, S.; Ye, L.; Li, Z.; Yang, C.; Pan, Q.; Liu, W.J.; Liu, H.-f. Mechanisms and functions of mitophagy and potential roles in renal disease. Front. Physiol. 2020, 11, 935.
- Sekine, S.; Youle, R.J. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2.
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550.
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119.
- Lu, S.; Liao, Z.; Lu, X.; Katschinski, D.M.; Mercola, M.; Chen, J.; Brown, J.H.; Molkentin, J.D.; Bossuyt, J.; Bers, D.M. Hyperglycemia Acutely Increases Cytosolic Reactive Oxygen Species via O-linked GlcNAcylation and CaMKII Activation in Mouse Ventricular Myocytes. Circ. Res. 2020, 126, e80–e96.
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070.
- King, G.L.; Loeken, M.R. Hyperglycemia-induced oxidative stress in diabetic complications. Histochem. Cell Biol. 2004, 122, 333–338.
- Hegarty, B.D.; Cooney, G.J.; Kraegen, E.W.; Furler, S.M. Increased efficiency of fatty acid uptake contributes to lipid accumulation in skeletal muscle of high fat-fed insulin-resistant rats. Diabetes 2002, 51, 1477–1484.
- Bastie, C.C.; Hajri, T.; Drover, V.A.; Grimaldi, P.A.; Abumrad, N.A. CD36 in myocytes channels fatty acids to a lipase-accessible triglyceride pool that is related to cell lipid and insulin responsiveness. Diabetes 2004, 53, 2209–2216.
- Bachmann, O.P.; Dahl, D.B.; Brechtel, K.; Machann, J.; Haap, M.; Maier, T.; Loviscach, M.; Stumvoll, M.; Claussen, C.D.; Schick, F. Effects of intravenous and dietary lipid challenge on intramyocellular lipid content and the relation with insulin sensitivity in humans. Diabetes 2001, 50, 2579–2584.
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800.
- Schrauwen, P.; Schrauwen-Hinderling, V.; Hoeks, J.; Hesselink, M.K. Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 266–271.
- Filippi, B.M.; Abraham, M.A.; Silva, P.N.; Rasti, M.; LaPierre, M.P.; Bauer, P.V.; Rocheleau, J.V.; Lam, T.K.T. Dynamin-Related Protein 1-Dependent Mitochondrial Fission Changes in the Dorsal Vagal Complex Regulate Insulin Action. Cell Rep. 2017, 18, 2301–2309.
- Serra, D.; Mera, P.; Malandrino, M.I.; Mir, J.F.; Herrero, L. Mitochondrial fatty acid oxidation in obesity. Antioxid. Redox Signal. 2013, 19, 269–284.
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 2004, 40, 185–194.
- Noeman, S.A.; Hamooda, H.E.; Baalash, A.A. Biochemical study of oxidative stress markers in the liver, kidney and heart of high fat diet induced obesity in rats. Diabetol. Metab. Syndr. 2011, 3, 17.
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658.
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950.
- Paltauf-Doburzynska, J.; Malli, R.; Graier, W.F. Hyperglycemic conditions affect shape and Ca2+ homeostasis of mitochondria in endothelial cells. J. Cardiovasc. Pharm. 2004, 44, 423–436.
- Vanhorebeek, I.; De Vos, R.; Mesotten, D.; Wouters, P.J.; De Wolf-Peeters, C.; Van den Berghe, G. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet 2005, 365, 53–59.
- Quirós, P.M.; Ramsay, A.J.; Sala, D.; Fernández-Vizarra, E.; Rodríguez, F.; Peinado, J.R.; Fernández-García, M.S.; Vega, J.A.; Enríquez, J.A.; Zorzano, A.; et al. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012, 31, 2117–2133.
- Sebastián, D.; Hernández-Alvarez, M.I.; Segalés, J.; Sorianello, E.; Muñoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528.
- Wang, L.; Ishihara, T.; Ibayashi, Y.; Tatsushima, K.; Setoyama, D.; Hanada, Y.; Takeichi, Y.; Sakamoto, S.; Yokota, S.; Mihara, K.; et al. Disruption of mitochondrial fission in the liver protects mice from diet-induced obesity and metabolic deterioration. Diabetologia 2015, 58, 2371–2380.
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645.
- Diaz-Morales, N.; Rovira-Llopis, S.; Bañuls, C.; Escribano-Lopez, I.; de Marañon, A.M.; Lopez-Domenech, S.; Orden, S.; Roldan-Torres, I.; Alvarez, A.; Veses, S.; et al. Are Mitochondrial Fusion and Fission Impaired in Leukocytes of Type 2 Diabetic Patients? Antioxid. Redox Signal. 2016, 25, 108–115.
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453.
- Finocchietto, P.; Perez, H.; Blanco, G.; Miksztowicz, V.; Marotte, C.; Morales, C.; Peralta, J.; Berg, G.; Poderoso, C.; Poderoso, J.J.; et al. Inhibition of mitochondrial fission by Drp-1 blockade by short-term leptin and Mdivi-1 treatment improves white adipose tissue abnormalities in obesity and diabetes. Pharm. Res. 2022, 178, 106028.
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319.
- Peng, L.; Men, X.; Zhang, W.; Wang, H.; Xu, S.; Fang, Q.; Liu, H.; Yang, W.; Lou, J. Involvement of dynamin-related protein 1 in free fatty acid-induced INS-1-derived cell apoptosis. PLoS ONE 2012, 7, e49258.
- Huang, M.; Wei, R.; Wang, Y.; Su, T.; Li, P.; Chen, X. The uremic toxin hippurate promotes endothelial dysfunction via the activation of Drp1-mediated mitochondrial fission. Redox Biol. 2018, 16, 303–313.
- Gao, L.; Liu, Y.; Wang, Y.; Chen, W.; Yang, K.; Li, J.; Lv, B.; Zhang, X.; Chi, J.; Liu, N.; et al. H2 relaxin ameliorates angiotensin II-induced endothelial dysfunction through inhibition of excessive mitochondrial fission. Biochem Biophys. Res. Commun. 2019, 512, 799–805.
- Zhang, B.; Guo, X.; Li, Y.; Peng, Q.; Gao, J.; Liu, B.; Wang, M. d-Chiro inositol ameliorates endothelial dysfunction via inhibition of oxidative stress and mitochondrial fission. Mol. Nutr. Food Res. 2017, 61, 1600710.
- Li, J.; Wang, Y.; Wang, Y.; Wen, X.; Ma, X.N.; Chen, W.; Huang, F.; Kou, J.; Qi, L.W.; Liu, B.; et al. Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. J. Mol. Cell. Cardiol. 2015, 86, 62–74.
- Li, Y.; Zhou, Z.H.; Chen, M.H.; Yang, J.; Leng, J.; Cao, G.S.; Xin, G.Z.; Liu, L.F.; Kou, J.P.; Liu, B.L.; et al. Inhibition of Mitochondrial Fission and NOX2 Expression Prevent NLRP3 Inflammasome Activation in the Endothelium: The Role of Corosolic Acid Action in the Amelioration of Endothelial Dysfunction. Antioxid. Redox Signal. 2016, 24, 893–908.
- Rogers, M.A.; Maldonado, N.; Hutcheson, J.D.; Goettsch, C.; Goto, S.; Yamada, I.; Faits, T.; Sesaki, H.; Aikawa, M.; Aikawa, E. Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ. Res. 2017, 121, 220–233.
- Ning, R.; Li, Y.; Du, Z.; Li, T.; Sun, Q.; Lin, L.; Xu, Q.; Duan, J.; Sun, Z. The mitochondria-targeted antioxidant MitoQ attenuated PM(2.5)-induced vascular fibrosis via regulating mitophagy. Redox Biol. 2021, 46, 102113.
- Abhijit, S.; Bhaskaran, R.; Narayanasamy, A.; Chakroborty, A.; Manickam, N.; Dixit, M.; Mohan, V.; Balasubramanyam, M. Hyperinsulinemia-induced vascular smooth muscle cell (VSMC) migration and proliferation is mediated by converging mechanisms of mitochondrial dysfunction and oxidative stress. Mol. Cell. Biochem. 2013, 373, 95–105.
- Zhang, X.; Chen, W.; Li, J.; Qi, S.; Hong, S.; Wang, Y.; Gao, L.; Shi, Z.; Liu, Y.; Liu, W.; et al. Involvement of mitochondrial fission in calcium sensing receptor-mediated vascular smooth muscle cells proliferation during hypertension. Biochem. Biophys. Res. Commun. 2018, 495, 454–460.
- Wu, Q.; Chen, Y.; Wang, Z.; Cai, X.; Che, Y.; Zheng, S.; Yuan, S.; Zhong, X. Mangiferin Inhibits PDGF-BB-Induced Proliferation and Migration of Rat Vascular Smooth Muscle Cells and Alleviates Neointimal Formation in Mice through the AMPK/Drp1 Axis. Oxid. Med. Cell. Longev. 2021, 2021, 3119953.
- Zhuang, X.; Maimaitijiang, A.; Li, Y.; Shi, H.; Jiang, X. Salidroside inhibits high-glucose induced proliferation of vascular smooth muscle cells via inhibiting mitochondrial fission and oxidative stress. Exp. Ther. Med. 2017, 14, 515–524.
- Wang, Q.; Zhang, M.; Torres, G.; Wu, S.; Ouyang, C.; Xie, Z.; Zou, M.-H. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes 2017, 66, 193–205.
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200.
- Zhong, Q.; Kowluru, R.A. Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8739–8746.
- Ding, M.; Dong, Q.; Liu, Z.; Liu, Z.; Qu, Y.; Li, X.; Huo, C.; Jia, X.; Fu, F.; Wang, X. Inhibition of dynamin-related protein 1 protects against myocardial ischemia–reperfusion injury in diabetic mice. Cardiovasc. Diabetol. 2017, 16, 19.
- Pegadraju, H.; Abby Thomas, J.; Kumar, R. Mechanistic and therapeutic role of Drp1 in the pathogenesis of stroke. Gene 2023, 855, 147130.
- Wu, Q.; Liu, J.; Mao, Z.; Tian, L.; Wang, N.; Wang, G.; Wang, Y.; Seto, S. Ligustilide attenuates ischemic stroke injury by promoting Drp1-mediated mitochondrial fission via activation of AMPK. Phytomedicine 2022, 95, 153884.
- Zhang, M.Y.; Zhu, L.; Zheng, X.; Xie, T.H.; Wang, W.; Zou, J.; Li, Y.; Li, H.Y.; Cai, J.; Gu, S.; et al. TGR5 Activation Ameliorates Mitochondrial Homeostasis via Regulating the PKCδ/Drp1-HK2 Signaling in Diabetic Retinopathy. Front. Cell Dev. Biol. 2021, 9, 759421.
- Zhang, M.Y.; Zhu, L.; Bao, X.; Xie, T.H.; Cai, J.; Zou, J.; Wang, W.; Gu, S.; Li, Y.; Li, H.Y.; et al. Inhibition of Drp1 ameliorates diabetic retinopathy by regulating mitochondrial homeostasis. Exp. Eye Res. 2022, 220, 109095.
- Chen, X.; Liang, J.; Bin, W.; Luo, H.; Yang, X. Anti-hyperlipidemic, Anti-inflammatory, and Ameliorative Effects of DRP1 Inhibition in Rats with Experimentally Induced Myocardial Infarction. Cardiovasc. Toxicol. 2021, 21, 1000–1011.
- Liu, D.; Zou, S.; Li, G.; Zhang, Q.; Chen, C.; Li, C.; Song, H.; Chen, S.; Wang, J.; Wu, Y.; et al. Downregulation of Uncoupling Protein 2(UCP2) Mediated by MicroRNA-762 Confers Cardioprotection and Participates in the Regulation of Dynamic Mitochondrial Homeostasis of Dynamin Related Protein1 (DRP1) After Myocardial Infarction in Mice. Front. Cardiovasc. Med. 2021, 8, 764064.
- Ding, J.; Zhang, Z.; Li, S.; Wang, W.; Du, T.; Fang, Q.; Wang, Y.; Wang, D.W. Mdivi-1 alleviates cardiac fibrosis post myocardial infarction at infarcted border zone, possibly via inhibition of Drp1-Activated mitochondrial fission and oxidative stress. Arch. Biochem. Biophys. 2022, 718, 109147.
- Xu, Y.; Wang, Y.; Wang, G.; Ye, X.; Zhang, J.; Cao, G.; Zhao, Y.; Gao, Z.; Zhang, Y.; Yu, B.; et al. YiQiFuMai Powder Injection Protects against Ischemic Stroke via Inhibiting Neuronal Apoptosis and PKCδ/Drp1-Mediated Excessive Mitochondrial Fission. Oxid. Med. Cell. Longev. 2017, 2017, 1832093.
- Kim, D.; Sesaki, H.; Roy, S. Reduced Levels of Drp1 Protect against Development of Retinal Vascular Lesions in Diabetic Retinopathy. Cells 2021, 10, 1379.
- Zhou, W.; Zhang, Y.; Jiao, Y.; Yin, W.; Dong, H.; Xu, S.; Tang, D.; Jiang, J.; Shao, J.; Wang, Z.; et al. Dexmedetomidine maintains blood-brain barrier integrity by inhibiting Drp1-related endothelial mitochondrial dysfunction in ischemic stroke. Acta Biochim. Biophys. Sin. (Shanghai) 2021, 53, 1177–1188.
- Chen, M.; Zhang, Q.; Wang, S.; Zheng, F. Inhibition of diabetes-induced Drp1 deSUMOylation prevents retinal vascular lesions associated with diabetic retinopathy. Exp. Eye Res. 2023, 226, 109334.
- Yang, D.Y.; Zhou, X.; Liu, Z.W.; Xu, X.Q.; Liu, C. LncRNA NEAT1 accelerates renal tubular epithelial cell damage by modulating mitophagy via miR-150-5p-DRP1 axis in diabetic nephropathy. Exp. Physiol. 2021, 106, 1631–1642.
- Chang, L.T.; Sun, C.K.; Wang, C.Y.; Youssef, A.A.; Wu, C.J.; Chua, S.; Yip, H.K. Downregulation of peroxisme proliferator activated receptor gamma co-activator 1alpha in diabetic rats. Int. Heart J. 2006, 47, 901–910.
- Zhang, Z.; Zhang, X.; Meng, L.; Gong, M.; Li, J.; Shi, W.; Qiu, J.; Yang, Y.; Zhao, J.; Suo, Y.; et al. Pioglitazone Inhibits Diabetes-Induced Atrial Mitochondrial Oxidative Stress and Improves Mitochondrial Biogenesis, Dynamics, and Function Through the PPAR-γ/PGC-1α Signaling Pathway. Front. Pharmcol. 2021, 12, 658362.
- Edwards, J.L.; Quattrini, A.; Lentz, S.I.; Figueroa-Romero, C.; Cerri, F.; Backus, C.; Hong, Y.; Feldman, E.L. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia 2010, 53, 160–169.
- Heinonen, S.; Buzkova, J.; Muniandy, M.; Kaksonen, R.; Ollikainen, M.; Ismail, K.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Vuolteenaho, K.; et al. Impaired Mitochondrial Biogenesis in Adipose Tissue in Acquired Obesity. Diabetes 2015, 64, 3135–3145.
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca2+ and AMPK/SIRT1. Nature 2010, 464, 1313–1319.
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124.
- Semple, R.K.; Crowley, V.C.; Sewter, C.P.; Laudes, M.; Christodoulides, C.; Considine, R.V.; Vidal-Puig, A.; O’Rahilly, S. Expression of the thermogenic nuclear hormone receptor coactivator PGC-1alpha is reduced in the adipose tissue of morbidly obese subjects. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 176–179.
- Roden, M. Muscle triglycerides and mitochondrial function: Possible mechanisms for the development of type 2 diabetes. Int. J. Obes. 2005, 29 (Suppl. S2), S111–S115.
- Kelley, D.E.; Goodpaster, B.; Wing, R.R.; Simoneau, J.A. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiol. 1999, 277, E1130–E1141.
- Simoneau, J.A.; Veerkamp, J.H.; Turcotte, L.P.; Kelley, D.E. Markers of capacity to utilize fatty acids in human skeletal muscle: Relation to insulin resistance and obesity and effects of weight loss. FASEB J. 1999, 13, 2051–2060.
- González-García, I.; Le Thuc, O.; Jastroch, M.; García-Cáceres, C. Divide et impera: How mitochondrial fission in astrocytes rules obesity. Mol. Metab. 2021, 45, 101159.
- Toda, C.; Kim, J.D.; Impellizzeri, D.; Cuzzocrea, S.; Liu, Z.W.; Diano, S. UCP2 Regulates Mitochondrial Fission and Ventromedial Nucleus Control of Glucose Responsiveness. Cell 2016, 164, 872–883.
- Ramírez, S.; Gómez-Valadés, A.G.; Schneeberger, M.; Varela, L.; Haddad-Tóvolli, R.; Altirriba, J.; Noguera, E.; Drougard, A.; Flores-Martínez, Á.; Imbernón, M.; et al. Mitochondrial Dynamics Mediated by Mitofusin 1 Is Required for POMC Neuron Glucose-Sensing and Insulin Release Control. Cell Metab. 2017, 25, 1390–1399.e1396.
- Dietrich, M.O.; Liu, Z.W.; Horvath, T.L. Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell 2013, 155, 188–199.
- Schneeberger, M.; Dietrich, M.O.; Sebastián, D.; Imbernón, M.; Castaño, C.; Garcia, A.; Esteban, Y.; Gonzalez-Franquesa, A.; Rodríguez, I.C.; Bortolozzi, A.; et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 2013, 155, 172–187.
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506.
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277.
- Bruce, C.R.; Risis, S.; Babb, J.R.; Yang, C.; Kowalski, G.M.; Selathurai, A.; Lee-Young, R.S.; Weir, J.M.; Yoshioka, K.; Takuwa, Y.; et al. Overexpression of sphingosine kinase 1 prevents ceramide accumulation and ameliorates muscle insulin resistance in high-fat diet-fed mice. Diabetes 2012, 61, 3148–3155.
- Schmitz-Peiffer, C.; Craig, D.L.; Biden, T.J. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J. Biol. Chem. 1999, 274, 24202–24210.
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15.
- Gutiérrez-Rodelo, C.; Roura-Guiberna, A.; Olivares-Reyes, J.A. Molecular Mechanisms of Insulin Resistance: An Update. Gac. Med. Mex. 2017, 153, 214–228.
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387.
- Komatsu, M.; Takei, M.; Ishii, H.; Sato, Y. Glucose-stimulated insulin secretion: A newer perspective. J. Diabetes Investig. 2013, 4, 511–516.
- Lin, H.-Y.; Weng, S.-W.; Chang, Y.-H.; Su, Y.-J.; Chang, C.-M.; Tsai, C.-J.; Shen, F.-C.; Chuang, J.-H.; Lin, T.-K.; Liou, C.-W.; et al. The Causal Role of Mitochondrial Dynamics in Regulating Insulin Resistance in Diabetes: Link through Mitochondrial Reactive Oxygen Species. Oxidative Med. Cell. Longev. 2018, 2018, 7514383.
- Chang, Y.H.; Lin, H.Y.; Shen, F.C.; Su, Y.J.; Chuang, J.H.; Lin, T.K.; Liou, C.W.; Lin, C.Y.; Weng, S.W.; Wang, P.W. The Causal Role of Mitochondrial Dynamics in Regulating Innate Immunity in Diabetes. Front. Endocrinol. 2020, 11, 445.
- Del Campo, A.; Parra, V.; Vásquez-Trincado, C.; Gutiérrez, T.; Morales, P.E.; López-Crisosto, C.; Bravo-Sagua, R.; Navarro-Marquez, M.F.; Verdejo, H.E.; Contreras-Ferrat, A.; et al. Mitochondrial fragmentation impairs insulin-dependent glucose uptake by modulating Akt activity through mitochondrial Ca2+ uptake. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1–E13.
- Dietrich, M.O.; Horvath, T.L. Feeding signals and brain circuitry. Eur. J. Neurosci. 2009, 30, 1688–1696.
- Broberger, C.; Johansen, J.; Johansson, C.; Schalling, M.; Hökfelt, T. The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc. Natl. Acad. Sci. USA 1998, 95, 15043–15048.
- Ollmann, M.M.; Wilson, B.D.; Yang, Y.K.; Kerns, J.A.; Chen, Y.; Gantz, I.; Barsh, G.S. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 1997, 278, 135–138.
- Horvath, T.L.; Bechmann, I.; Naftolin, F.; Kalra, S.P.; Leranth, C. Heterogeneity in the neuropeptide Y-containing neurons of the rat arcuate nucleus: GABAergic and non-GABAergic subpopulations. Brain Res. 1997, 756, 283–286.
- Diano, S. New aspects of melanocortin signaling: A role for PRCP in α-MSH degradation. Front. Neuroendocr. 2011, 32, 70–83.
- McClellan, K.M.; Calver, A.R.; Tobet, S.A. GABAB receptors role in cell migration and positioning within the ventromedial nucleus of the hypothalamus. Neuroscience 2008, 151, 1119–1131.
- Hahn, T.M.; Breininger, J.F.; Baskin, D.G.; Schwartz, M.W. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat. Neurosci. 1998, 1, 271–272.
- Kohno, D.; Sone, H.; Minokoshi, Y.; Yada, T. Ghrelin raises i via AMPK in hypothalamic arcuate nucleus NPY neurons. Biochem. Biophys. Res. Commun. 2008, 366, 388–392.
- Sternson, S.M.; Shepherd, G.M.; Friedman, J.M. Topographic mapping of VMH --> arcuate nucleus microcircuits and their reorganization by fasting. Nat. Neurosci. 2005, 8, 1356–1363.
- Takahashi, K.A.; Cone, R.D. Fasting induces a large, leptin-dependent increase in the intrinsic action potential frequency of orexigenic arcuate nucleus neuropeptide Y/Agouti-related protein neurons. Endocrinology 2005, 146, 1043–1047.
- Yang, Y.; Atasoy, D.; Su, H.H.; Sternson, S.M. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 2011, 146, 992–1003.
- Cone, R.D. Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 2005, 8, 571–578.
- Büch, T.R.; Heling, D.; Damm, E.; Gudermann, T.; Breit, A. Pertussis toxin-sensitive signaling of melanocortin-4 receptors in hypothalamic GT1-7 cells defines agouti-related protein as a biased agonist. J. Biol. Chem. 2009, 284, 26411–26420.
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdán, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484.
- Horvath, T.L.; Naftolin, F.; Kalra, S.P.; Leranth, C. Neuropeptide-Y innervation of beta-endorphin-containing cells in the rat mediobasal hypothalamus: A light and electron microscopic double immunostaining analysis. Endocrinology 1992, 131, 2461–2467.
- Beck, B. Neuropeptide Y in normal eating and in genetic and dietary-induced obesity. Philos. Trans. R. Soc. Lond B Biol. Sci. 2006, 361, 1159–1185.
- Kokot, F.; Ficek, R. Effects of neuropeptide Y on appetite. Min. Electrolyte Metab. 1999, 25, 303–305.
- Marraudino, M.; Bo, E.; Carlini, E.; Farinetti, A.; Ponti, G.; Zanella, I.; Di Lorenzo, D.; Panzica, G.C.; Gotti, S. Hypothalamic Expression of Neuropeptide Y (NPY) and Pro-OpioMelanoCortin (POMC) in Adult Male Mice Is Affected by Chronic Exposure to Endocrine Disruptors. Metabolites 2021, 11, 368.
- Spanswick, D.; Smith, M.A.; Groppi, V.E.; Logan, S.D.; Ashford, M.L. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature 1997, 390, 521–525.
- Spanswick, D.; Smith, M.A.; Mirshamsi, S.; Routh, V.H.; Ashford, M.L. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat. Neurosci. 2000, 3, 757–758.
- Van den Top, M.; Lee, K.; Whyment, A.D.; Blanks, A.M.; Spanswick, D. Orexigen-sensitive NPY/AgRP pacemaker neurons in the hypothalamic arcuate nucleus. Nat. Neurosci. 2004, 7, 493–494.
- Claret, M.; Smith, M.A.; Batterham, R.L.; Selman, C.; Choudhury, A.I.; Fryer, L.G.; Clements, M.; Al-Qassab, H.; Heffron, H.; Xu, A.W.; et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J. Clin. Investig. 2007, 117, 2325–2336.
- Cowley, M.A.; Smith, R.G.; Diano, S.; Tschöp, M.; Pronchuk, N.; Grove, K.L.; Strasburger, C.J.; Bidlingmaier, M.; Esterman, M.; Heiman, M.L.; et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003, 37, 649–661.
- Chen, S.R.; Chen, H.; Zhou, J.J.; Pradhan, G.; Sun, Y.; Pan, H.L.; Li, D.P. Ghrelin receptors mediate ghrelin-induced excitation of agouti-related protein/neuropeptide Y but not pro-opiomelanocortin neurons. J. Neurochem. 2017, 142, 512–520.
- Méquinion, M.; Foldi, C.J.; Andrews, Z.B. The Ghrelin-AgRP Neuron Nexus in Anorexia Nervosa: Implications for Metabolic and Behavioral Adaptations. Front. Nutr. 2019, 6, 190.
- Vong, L.; Ye, C.; Yang, Z.; Choi, B.; Chua, S., Jr.; Lowell, B.B. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 2011, 71, 142–154.
- Varela, L.; Horvath, T.L. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep. 2012, 13, 1079–1086.
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Münzberg, H.; Zhang, Z.Y.; Kahn, B.B.; et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 2015, 160, 88–104.
- Dodd, G.T.; Tiganis, T. Insulin action in the brain: Roles in energy and glucose homeostasis. J. Neuroendocr. 2017, 29, e2513.
- Timper, K.; Brüning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Model Mech. 2017, 10, 679–689.
- Shulman, R.G.; Rothman, D.L.; Behar, K.L.; Hyder, F. Energetic basis of brain activity: Implications for neuroimaging. Trends Neurosci. 2004, 27, 489–495.
- Nasrallah, C.M.; Horvath, T.L. Mitochondrial dynamics in the central regulation of metabolism. Nat. Rev. Endocrinol. 2014, 10, 650–658.
- Gómez-Valadés, A.G.; Pozo, M.; Varela, L.; Boudjadja, M.B.; Ramírez, S.; Chivite, I.; Eyre, E.; Haddad-Tóvolli, R.; Obri, A.; Milà-Guasch, M.; et al. Mitochondrial cristae-remodeling protein OPA1 in POMC neurons couples Ca(2+) homeostasis with adipose tissue lipolysis. Cell Metab. 2021, 33, 1820–1835.e1829.
- Santoro, A.; Campolo, M.; Liu, C.; Sesaki, H.; Meli, R.; Liu, Z.W.; Kim, J.D.; Diano, S. DRP1 Suppresses Leptin and Glucose Sensing of POMC Neurons. Cell Metab. 2017, 25, 647–660.
- Jin, S.; Diano, S. Mitochondrial Dynamics and Hypothalamic Regulation of Metabolism. Endocrinology 2018, 159, 3596–3604.
- Patel, B.; New, L.E.; Griffiths, J.C.; Deuchars, J.; Filippi, B.M. Inhibition of mitochondrial fission and iNOS in the dorsal vagal complex protects from overeating and weight gain. Mol. Metab. 2021, 43, 101123.
- Chang, C.R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39.
- Bo, T.; Yamamori, T.; Suzuki, M.; Sakai, Y.; Yamamoto, K.; Inanami, O. Calmodulin-dependent protein kinase II (CaMKII) mediates radiation-induced mitochondrial fission by regulating the phosphorylation of dynamin-related protein 1 (Drp1) at serine 616. Biochem. Biophys. Res. Commun. 2018, 495, 1601–1607.
- Kashatus, D.F.; Lim, K.-H.; Brady, D.C.; Pershing, N.L.; Cox, A.D.; Counter, C.M. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell Biol. 2011, 13, 1108–1115.
- Merz, K.E.; Hwang, J.; Zhou, C.; Veluthakal, R.; McCown, E.M.; Hamilton, A.; Oh, E.; Dai, W.; Fueger, P.T.; Jiang, L.; et al. Enrichment of the exocytosis protein STX4 in skeletal muscle remediates peripheral insulin resistance and alters mitochondrial dynamics via Drp1. Nat. Commun. 2022, 13, 424.
- Civitarese, A.E.; Carling, S.; Heilbronn, L.K.; Hulver, M.H.; Ukropcova, B.; Deutsch, W.A.; Smith, S.R.; Ravussin, E. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007, 4, e76.
- Mesquita, P.H.C.; Vann, C.G.; Phillips, S.M.; McKendry, J.; Young, K.C.; Kavazis, A.N.; Roberts, M.D. Skeletal Muscle Ribosome and Mitochondrial Biogenesis in Response to Different Exercise Training Modalities. Front. Physiol. 2021, 12, 725866.
- Viña, J.; Gomez-Cabrera, M.C.; Borras, C.; Froio, T.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Pallardo, F.V. Mitochondrial biogenesis in exercise and in ageing. Adv. Drug Deliv. Rev. 2009, 61, 1369–1374.
- Holloszy, J.O. Regulation of mitochondrial biogenesis and GLUT4 expression by exercise. Compr. Physiol. 2011, 1, 921–940.
- Hood, D.A.; Uguccioni, G.; Vainshtein, A.; D’Souza, D. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle: Implications for health and disease. Compr. Physiol. 2011, 1, 1119–1134.
- Pengam, M.; Amérand, A.; Simon, B.; Guernec, A.; Inizan, M.; Moisan, C. How do exercise training variables stimulate processes related to mitochondrial biogenesis in slow and fast trout muscle fibres? Exp. Physiol. 2021, 106, 938–957.
- Nishio, Y.; Kanazawa, A.; Nagai, Y.; Inagaki, H.; Kashiwagi, A. Regulation and role of the mitochondrial transcription factor in the diabetic rat heart. Ann. N. Y. Acad. Sci. 2004, 1011, 78–85.
- Hood, D.A. Invited Review: Contractile activity-induced mitochondrial biogenesis in skeletal muscle. J. Appl. Physiol (1985) 2001, 90, 1137–1157.
- Dillon, L.M.; Rebelo, A.P.; Moraes, C.T. The role of PGC-1 coactivators in aging skeletal muscle and heart. IUBMB Life 2012, 64, 231–241.
- Wenz, T.; Rossi, S.G.; Rotundo, R.L.; Spiegelman, B.M.; Moraes, C.T. Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. USA 2016, 113, E8502.
- Wei, P.; Guo, J.; Xue, W.; Zhao, Y.; Yang, J.; Wang, J. RNF34 modulates the mitochondrial biogenesis and exercise capacity in muscle and lipid metabolism through ubiquitination of PGC-1 in Drosophila. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 1038–1046.
- Egan, B.; Carson, B.P.; Garcia-Roves, P.M.; Chibalin, A.V.; Sarsfield, F.M.; Barron, N.; McCaffrey, N.; Moyna, N.M.; Zierath, J.R.; O’Gorman, D.J. Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor coactivator-1 mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J. Physiol. 2010, 588, 1779–1790.
- Edgett, B.A.; Foster, W.S.; Hankinson, P.B.; Simpson, C.A.; Little, J.P.; Graham, R.B.; Gurd, B.J. Dissociation of increases in PGC-1α and its regulators from exercise intensity and muscle activation following acute exercise. PLoS ONE 2013, 8, e71623.
- Song, K.; Zhang, Y.; Ga, Q.; Bai, Z.; Ge, R.L. Increased Insulin Sensitivity by High-Altitude Hypoxia in Mice with High-Fat Diet-Induced Obesity Is Associated with Activated AMPK Signaling and Subsequently Enhanced Mitochondrial Biogenesis in Skeletal Muscles. Obes. Facts 2020, 13, 455–472.
- Short, K.R.; Vittone, J.L.; Bigelow, M.L.; Proctor, D.N.; Rizza, R.A.; Coenen-Schimke, J.M.; Nair, K.S. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes 2003, 52, 1888–1896.
- Russell, A.P.; Feilchenfeldt, J.; Schreiber, S.; Praz, M.; Crettenand, A.; Gobelet, C.; Meier, C.A.; Bell, D.R.; Kralli, A.; Giacobino, J.P.; et al. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes 2003, 52, 2874–2881.
- Garnier, A.; Fortin, D.; Zoll, J.; N’Guessan, B.; Mettauer, B.; Lampert, E.; Veksler, V.; Ventura-Clapier, R. Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle. FASEB J. 2005, 19, 43–52.
- Baar, K.; Wende, A.R.; Jones, T.E.; Marison, M.; Nolte, L.A.; Chen, M.; Kelly, D.P.; Holloszy, J.O. Adaptations of skeletal muscle to exercise: Rapid increase in the transcriptional coactivator PGC-1. FASEB J. 2002, 16, 1879–1886.
- Pilegaard, H.; Saltin, B.; Neufer, P.D. Exercise induces Transient Transcriptional Activation of the PGC-1α Gene in Human Skeletal Muscle; Wiley Online Library: Hoboken, NJ, USA, 2003.
This entry is offline, you can click here to edit this entry!