Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Mitochondria play a major role in ROS production and defense during their life cycle. The transcriptional activator PGC-1α is a key player in the homeostasis of energy metabolism and is therefore closely linked to mitochondrial function. PGC-1α responds to environmental and intracellular conditions and is regulated by SIRT1/3, TFAM, and AMPK, which are also important regulators of mitochondrial biogenesis and function.
- PGC-1α
- ROS defense
- mitonuclear communication
1. Introduction
The peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α is described as the master regulator of mitochondrial biogenesis and function. It was identified together with the peroxisome proliferator-activated receptor γ (PPARγ) transcription factor in mitochondria-rich and thermogenesis-specialized brown adipose tissue (BAT). Current studies suggest a role of PGC-1α in the regulation of oxidative phosphorylation (OXPHOS), fatty acid (FA)/lipid metabolism, and the modulation of reactive oxygen species (ROS). PGC-1α is found mainly in metabolically active tissues, such as the liver, kidney, skeletal muscle, brain, and adipose tissue [1,2,3]. It is also involved in the transformation of white adipose tissue into brown adipose tissue [4]. In mammals, fasting, exercise, and cold are associated with an increase in PGC-1α levels [5,6,7]. PGC-1α subsequently upregulates respiratory gene expression in the mitochondria [8].
PGC-1α belongs to the so-called PGC family of transcriptional regulators. Other members of the family are PGC-1β and PRC (PGC-1-related coactivator). The human PGC-1α consists of 798 amino acids, has a molecular weight of 91 kDa, and can be divided into several functional regions, such as the activation domain, inactivation domain, short serine/arginine-rich stretches (RS) domain, and the RNA recognition motif (RRM) [7,9] (Figure 1A). Its structure is not resolved yet.
PGC-1 family members do not have a DNA-binding domain. Also, PGC-1α does not have an intrinsic histone acetyltransferase activity, which is present in other transcriptional coactivators, that initiates gene transcription and chromatin remodeling. PGC-1α acts more as a transcriptional regulator by providing a docking platform for proteins that possess histone acetyltransferase activity. Therefore, PGC-1α indirectly promotes transcription [10,11].
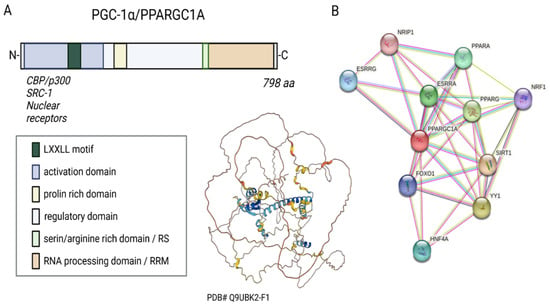
Figure 1. Sequence, putative structure, and interactions of PGC-1α. (A) Domains and structure of PGC-1α. Structure generated with α-fold. (B) Known and experimental interactions of the PGC-1α/PPARGC with proteins based on the STRING database entries.
Three LXXLL leucine-rich motifs (NR boxes) are located at the N-terminal of PGC-1α’s activation region and its adjacent inactive region. These can bind to several nuclear receptors, such as NR, peroxisome proliferator-activated receptor α (PPARα), estrogen receptor (ER), or nuclear respiratory factor 1 and 2 (NRF-1/NRF-2) (Figure 1B) [14,15,16]. The serine/arginine-rich domain (RS) and the RNA processing domain (RRM) motifs towards the C-terminus are typical for proteins involved in RNA splicing [17,18]. Monsalve et al. showed in in vitro studies that the C-terminal functional region participates in mRNA(messenger RNA) processing to regulate gene expression. Mutations in the RS and RRM motifs of PGC-1α affect PGC-1α’s ability to interact with transcription factors and thus impair gene transcription [19].
2. Regulation of PGC-1α
2.1. Splice Variants of PGC-1α
PGC-1α gene transcription is regulated by multiple promoter regions and is coupled with alternative splicing, resulting in a variety of PGC-1α protein variants [20]. The combination of alternative splicing and alternative use of promoters is a common process for increasing transcriptome complexity [21]. New splice variants are generated by transcription from an evolutionarily conserved alternative promoter (AP), which was found approximately 14 kb upstream of PGC-1α’s transcription start site (TSS) [22]. The transcripts of this AP contain a new exon 1 (exon 1b) with two splicing options, resulting in proteins with two different amino termini (PGC-1α-b, 12 aa long and PGC-1α-c, 3 aa long). This exon is shorter than the proximal exon 1a, which encodes for a 16 aa-long N-terminus [22,23]. The newly discovered isoforms were found in skeletal muscle after exercise [24,25,26,27] and apparently, their generation is more responsive to stimulation. Studies with PGC-1α-b in the skeletal muscle of transgenic mice revealed that a change in the mitochondrial volume is directly correlated with an improvement in exercise performance and oxidative capacity [28]. Nevertheless, the structure and function of PGC-1α-b and PGC-1α-c do not differ notably from the canonical PGC-1α [22,23].
In addition to the AP and its resulting isoforms, further TSSs and tissue-specific promoters have been described for PGC-1α in the liver, kidney, and brain. These isoforms need to be studied in a tissue-specific context and may vary functionally and structurally from the canonical PGC-1α [29,30,31].
2.2. Regulation of the Master Regulator
PGC-1α is tightly regulated at two different levels: Firstly, PGC-1α is regulated at the transcriptional level (see also Section 2.1) through several transcription factors and various extracellular stimuli such as insulin/glucagon levels, Ca2+, temperature, or exercise via signaling cascades. Secondly, PGC-1α is regulated at the posttranslational level through numerous modifications, such as acetylation, phosphorylation, methylation, or ubiquitination [32,33]. The overview in Figure 2 shows the most important signaling routes that target PGC-1α through posttranslational and transcriptional modifications.
2.3. Stress-Related Transcriptional Regulation of PGC-1α
Various factors play a role in the regulation of PGC-1α, e.g., the transcription of PGC-1α is upregulated by forkhead box class-01 (FoxO1), myocyte enhancer factor 2 (MEF2), activating transcription factor 2 (ATF2), and cyclic AMP response element-binding protein (CREB). These upstream factors are induced by several extracellular stimuli such as stress, exercise, or cytokines [32,34]. Early-phase mediators of inflammation, such as the tumor necrosis factor α (TNFα), interleukin-4, and interferon-γ, regulate PGC-1α gene expression. Nuclear factor-kappa B (NF-κB), mitogen-activated protein kinase (MAPK), or Akt serine/threonine kinase (protein kinase B; Akt; PKB) have a mediating effect on these signaling pathways as well [35].
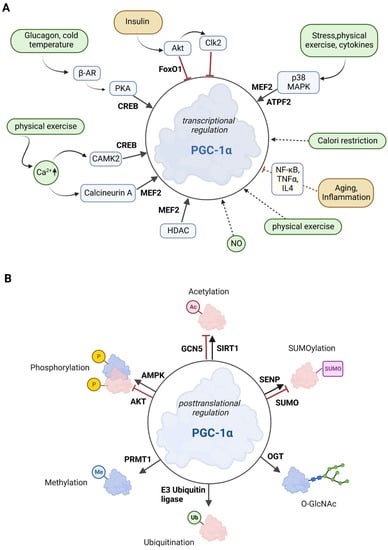
Figure 2. Overview of the effectors of PGC-1α. (A) Posttranscriptional control of PGC-1α. The upregulators are in green boxes, while the downregulators are shown in yellow color. Transcription factors that control PGC-1α gene expression are printed in bold. The posttranslational mediators are in light blue. (B) Posttranslational modifications of PGC-1α. Several sites for modifications including phosphorylation, acetylation, methylation, ubiquitination, O-GlcNAcylation (O-linked N-acetyl glucosylation), and SUMOylation have been described for PGC-1α. Red molecules indicate inactivation, and blue indicates activation.
p38 MAPK activates MEF2 and ATF2, both of which stimulate PGC-1α expression. Exercise increases Ca2+ levels, leading to the activation of MEF and CREB factors via calcineurin A and Ca2+/calmodulin-dependent protein kinase 2 (CAMK2) [32,37]. Activation of 5′ adenosine monophosphate-activated protein kinase (AMPK) via CAMK2 is promoted by calcium ions [38]. FoxO1 and Akt are activated by insulin, whereas glucagon (via glucagon receptor) and cold temperature (via β3-adrenergic receptor; β-AR) stimulate protein kinase A (PKA), which subsequently promotes CREB-mediated transcription [32]. In summary, the combination of a variety of response factors, integrating environmental and intracellular stimuli, controls the PGC-1α gene expression [36].
Class II histone deacetylases (HDACs) inhibit the MEF2 transcription factor and indirectly regulate PGC-1α gene expression. In HDAC-negative mouse models, MEF2 activity was increased, resulting in enhanced skeletal muscle development (endurance and resistance to fatigue) [39]. This observation may be related to an increase in the PGC-1α expression [36].
Recruitment of RNA polymerase II to the PGC-1α promoter is inhibited by its phosphorylation by cyclin-dependent kinase 9 [40]. In addition, transcription factors EB (TFEB)and E3 (Tfe3), have been reported to directly regulate the PGC-1α gene [41].
In addition, various epigenetic modifications of the PGC-1α promoter regulate PGC-1α gene expression [42]. Barrès et al. showed that promoter methylation in muscle cells by DNA methyltransferase 3B (DNMT3B) leads to the repression of the PGC-1α gene in the presence of high levels of FA [43]. This is relevant with respect to the involvement of PGC-1α in the control of mitochondrial biogenesis and the regulation of mitophagy [33,44]. Aging and inflammation, which are often associated with an increase in ROS, also affect PGC-1α gene expression.
2.4. Posttranslational Regulation of PGC-1α
AMPK, MAPK, Akt, S6 kinase, and glycogen synthase kinase 3β (GSK3β) are the major and best-described protein kinases that target PGC-1α for posttranslational phosphorylation (Figure 2B) [32,45].
AMPK is activated when the cellular AMP/ATP ratio increases. It is, therefore, a key enzyme in mitochondrial energy homeostasis. Increased AMPK activity results in the inhibition of cell growth, proliferation, and anabolic processes such as lipid synthesis [46]. Specifically, AMPK binds to PGC-1α in muscle cells and phosphorylates Thr177 and Ser538. This phosphorylation increases the transcriptional activity of PGC-1α. Furthermore, these phosphorylations are required for AMPK-induced gene expression of mitochondrial genes, glucose transporter 4 (GLUT4), and PGC-1α itself [47]. In addition, increased protein stability is a result of the p38 MAPK-induced phosphorylation of PGC-1α at Thr262, Ser265, and Thr298 [32]. In conclusion, cellular energy balance is primarily regulated by the AMPK/PGC-1α axis, which largely controls mitochondrial energy metabolism. This balance can be disrupted by chronic overnutrition, which triggers the shutdown of AMPK expression and leads to impaired PGC-1α activity, resulting in mitochondrial dysfunction [15,47].
Akt is involved in several cellular signaling and regulatory pathways, such as PGC-1α regulation. It is known that phosphorylation can also decrease PGC-1α activity in cells; for example, Akt can phosphorylate several C-terminal sites of PGC-1α. By phosphorylation of PGC-1α, Akt inhibits both gluconeogenesis and fatty acid oxidation (FAO) [48]. Akt, activated in the liver upon feeding, phosphorylates PGC-1α at Ser568 and Ser572, which inhibits the gluconeogenic program of downstream targets. However, these specific phosphorylations do not affect the function of PGC-1α as an activator of mitochondrial and FAO genes [49]. Akt is also involved in the phosphate-3-kinase-Akt-mechanistic target of rapamycin (mechanistic target of rapamycin; mTOR) signaling pathway. This pathway controls several cellular mechanisms (survival, differentiation, growth, metabolism, and cancer) and inhibits the PGC-1α response [50,51]. In addition, Akt activates CDC2-like kinase 2 (Clk2), which also mediates the PGC-1α inactivation [52]. Akt also inhibits the PGC-1α-mediated activation of the FoxO1 transcription factor [32,53]. In 3T3 cells, the stability of PGC-1α is regulated by GSK3β, which targets PGC-1α for intranuclear proteasomal degradation [54].
Sirtuin 1 (SIRT1) belongs to the family of the silent information regulator 2-related histone deacetylase family [55]. To mediate the deacetylation of target substrates, sirtuin proteins require nicotinamide adenine dinucleotide (NAD) [56]. Since the cellular REDOX balance of NAD+ and NADH is closely linked to catabolic metabolism, it is proposed that SIRT1 acts as a sensor that directly links metabolic perturbations to transcriptional outputs. As such, SIRT1 interacts with PGC-1α and deacetylates it in an NAD+-dependent manner (Figure 2B) [57]. It has been suggested that PGC-1α and SIRT1 are mitochondrially imported proteins localized in the mitochondrial matrix [58], but the evidence suggest that SIRT1 is a nuclear/cytosolic protein [59], while SIRT3is located in the mitochondria [60]. SIRTs primarily affect mitochondrial function, with two existing pathways: a PGC-1α-dependent and a PGC-1α-independent pathway [15]. In the PGC-1α-dependent pathway, SIRT1 activates PGC-1α through deacetylation. Activated PGC-1α acts as a coactivator for mitochondrial transcription factor A (TFAM), which is thought to promote the transport of SIRT1 and PGC-1α into mitochondria where they form a complex with the D-loop region of mtDNA. The D-loop region regulates mitochondrial DNA replication and transcription [58,59]. SIRT1 activity can be enhanced by exercise and fasting [61,62]. Fasting has been shown to induce SIRT1-dependent PGC-1α deacetylation in skeletal muscle and is required for the activation of mitochondrial FAO proteins under low glucose conditions [63]. In contrast, histone acetyltransferase activity controls non-depressible 5 (GCN5), which results in PGC-1α acetylation. In addition, the SIRT1 inhibitor, nicotinamide, induces PGC-1α acetylation, thereby reducing the expression of PGC-1α target genes. Cellular energy overload leads to increased levels of steroid receptor coactivator 3 (SRC-3), resulting in GCN5 up-regulation and thus pronounced PGC-1α acetylation [55].
Specific ubiquitination (Ub) of PGC-1α by the E3 ubiquitin ligase SCFCdc4 (Skp1/Cullin/F-box cell division control 4) results in a very short half-life (0.3 h) of PGC-1α in the nucleus due to proteolytic digestion (Figure 2B) [64]. Conversely, decreased SCFCdc4 activity results in PGC-1α accumulation in response to oxidative stress, thus providing an increased ability to neutralize toxic metabolic byproducts such as ROS [65].
The small ubiquitin-like modifier (SUMO)-1 protein attenuates the activity of PGC-1α through SUMOylation [66]. SUMOylation of PGC-1α inactivates the enzyme, which is reversed by a Sentrin/SUMO-specific protease (SENP1) that de-SUMOylates PGC-1α and thus results in mitochondrial biogenesis [67].
Methylation by the protein arginine methyltransferase 1 (PRMT1) increases PGC-1α activity and induces the transcription of genes important for mitochondrial biogenesis [68] (Figure 2B).
Another posttranslational modification is O-GlcNAcylation, which is the addition of β-N-acetylglucosamine (GlcNAc) groups by O-linked β-N-acetylglucosamine (O-GlcNAc) transferase (OGT). This stabilizes PGC-1α by inhibiting its ubiquitination [69] (Figure 2B). In addition, O-GlcNAcylation of the transcription factor FoxO1 and the CREB-regulated transcription co-activator 2 (CRTC2) is associated with PGC-1α activity. During its interaction with PGC-1α, OGT transfers a GlcNAc group to FoxO1 and is then able to modify CRTC2. This is thought to be necessary for the interaction of CRTC2 with PGC-1α, resulting in increased PGC-1α gene expression. O-GlcNAcylation of specific transcription-related factors, such as PGC-1α and FoxO1, is important for nutrient stress sensing and cellular energy metabolism [70].
In summary, various posttranslational modifications create a versatile and efficient array for regulating the activity and intracellular localization of PGC-1α, thus ultimately contributing to the pivotal role of PGC-1α in mitochondrial energy metabolism and biogenesis [71].
3. The Link between PGC-1α and Mitochondria
3.1. PGC-1α as the Master Regulator of Mitochondrial Biogenesis
PGC-1α is the master regulator of mitochondrial biogenesis and an important regulator of mitochondrial oxidative capacity (Figure 3). This occurs through a variety of transcription factors, such as ERR, PPARγ, and NRF-1/2, which are coactivated by PGC-1α and all play an important role in mitochondrial oxidative capacity [72,73]. In addition, the interaction between PPARγ and PGC-1α can stimulate mitochondrial biogenesis through the regulation of PGC-1α activity itself. Specifically, PGC-1α and PPARγ control proteins involved in the regulation of mitochondrial biogenesis, including promoting OXPHOS gene expression in the nucleus and mitochondria and stimulating mtDNA replication, thereby enhancing mitochondrial function and metabolism [74,75,76]. The upstream gatekeepers of PGC-1α activity are SIRT1 and AMPK, which are important actuators in the regulatory network of metabolic homeostasis [77,78].
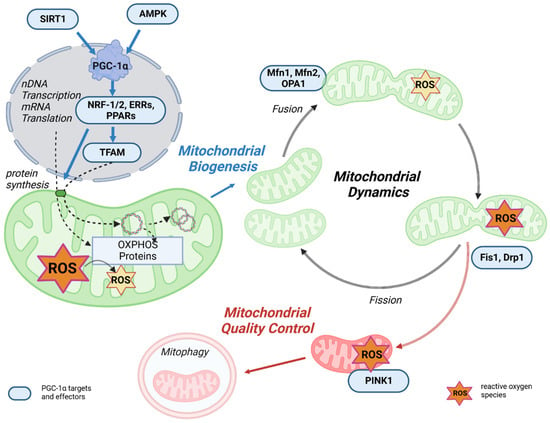
Figure 3. The link between mitochondrial life cycle and PGC-1α. Mitochondrial biogenesis is initiated by an energetic imbalance sensed by two pathways: AMPK and SIRT1. Increased expression or activity of the key regulator of mitochondrial biogenesis PGC-1α activates the expression of NRF-1/2, which induces the expression of TFAM, which translocates to mitochondria, binds to mtDNA, and activates transcription and replication. An increase in OXPHOS proteins reduces ROS generation in mitochondria. Mitochondrial fusion and fission dynamics are also affected by ROS. Dysfunctional mitochondria can be eliminated through a process known as mitophagy.
Mitochondrial transcription is activated by PGC-1α, PGC-1β, and PRC, but PGC-1α is the master regulator of mitochondrial biogenesis. The process is initiated when PGC-1α is activated by phosphorylation of AMPK or deacetylation of SIRT1 and stimulates various nuclear transcription factors, such as NRF-1, NRF-2, and estrogen-related receptor alpha (ERRα). Through activation of NRF-1/2 [80], PGC-1α promotes TFAM transcription and expression [81,82]. In addition, NRF-2 regulates the gene expression of the protein import receptor Tom70 (Tom70) of the translocase of the outer mitochondrial membrane (TOM) [83]. TFAM stimulates the transcription and replication of mtDNA [84,85], but the correlation between TFAM levels and mtDNA transcription and replication may be complex [86]. In the next step, specific translation factors, such as mtIF2 and mtIF3, translate the mtDNA. In terms of energy metabolism, the PGC-1α-NRF-1/2 pathway promotes the gene expression of mitochondrial complexes I, II, III, IV, and cytochrome c, thereby activating OXPHOS [87].
In summary, mitochondrial biogenesis must undergo mtDNA transcription and translation, demonstrating that upregulation of transcription factor activation via PGC-1α is a key step in mitochondrial biogenesis.
3.2. PGC-1α Affects Mitochondrial Dynamics and Quality Control
Complementary to mitochondrial biogenesis, mitochondrial quality control is a key process for maintaining the energy supply by mitochondria. Mitochondrial quality control is a multilevel process involving multiple mitochondrial and cytosolic proteases, protein replenishment, and mitophagy [88]. Maintenance of mitochondrial performance and adaptation to changing energy demands is regulated by remodeling mitochondrial structures, which is primarily controlled by fission/fusion, mitochondrial biogenesis, and mitophagy [89]. Intriguingly, in addition to its role in regulating mitochondrial biogenesis, PGC-1α is also involved in mitochondrial dynamics and mitophagy [90]. An overview of these processes is shown in Figure 3.
It is well known that mitochondria are dynamic organelles that continuously undergo the processes of fusion and fission. Mitochondrial fusion is controlled by mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) in the mitochondrial outer membrane and optic atrophy 1 (Opa1) in the mitochondrial inner membrane [91,92]. These fusion proteins contain functional GTPase domains and, upon activation, result in an expanded, branched mitochondrial network. Mitochondrial fission is the process that counteracts fusion and allows the mitochondrial network to split, resulting in small, fragmented, and globular mitochondria. Fission is also regulated by GTPase proteins, such as fission 1 protein (Fis1) and dynamin-related protein (Drp1) [93,94]. Healthy mitochondrial dynamics are regulated by maintaining a balance between these opposing processes, which is fundamental to maintaining mitochondrial quality and function [89]. According to Peng et al., it can be speculated that excessive mitochondrial fragmentations lead to mitochondrial dysfunction [95]. PGC-1α can slow down the fission of neuronal mitochondria by regulating Drp1 levels [96], resulting in increased mitochondrial fusion. This may prevent or slow down the damage and denaturation of neuronal axons caused by ATP depletion induced by mitochondrial fragmentation. Activation of PPARγ decreases Drp1 activity through phosphorylation, thereby reducing excessive mitochondrial fission and neuronal damage [97]. The beneficial effect of PPARγ/PGC-1α to reduce mitochondrial oxidative stress response via stimulation of mitochondrial biogenesis, dynamics, and function was also demonstrated in a rabbit model of diabetes associated with atrial ROS stress [98]. PGC-1α was shown to directly regulate Mfn1 gene transcription by coactivating the ERRα, which ultimately promotes mitochondrial fusion (Figure 3) [99]. In PGC-1α overexpression and knock-out cell models, PGC-1α was shown to regulate Mfn2 and p-Drp1 protein expression and phosphorylation [95], which are important for the balance between mitochondrial dynamics, function, and homeostasis. In summary, PGC-1α provides a link between mitochondrial biogenesis and fission/fusion [95].
Finally, mitochondrial degradation by autophagy is also mediated by PGC-1α through transcriptional mechanisms [100]. Autophagy is the major process by which damaged organelles and cellular by-products are degraded and recycled in the lysosome to maintain cellular homeostasis [101]. Mitophagy is the mitochondria-specific form of autophagy. Damaged and/or dysfunctional mitochondria are often characterized by a disturbance of the mitochondrial membrane potential, which may also have effects on the sensitive ROS balance [102]. Depolarization of mitochondria leads to the recruitment of PTEN-induced kinase 1 (PINK1), which activates parkin, an E3 ubiquitin ligase, followed by ubiquitination of outer membrane proteins. This mitochondria-ubiquitinated complex is then engulfed by the autophagosome and degraded in the lysosome. Mitophagy is critical for maintaining healthy mitochondria in various tissues and disease states by deleting defective mitochondrial segments within the network [89]. In addition, PGC-1α can interact with and stabilize the mRNA of mitostatin, a mitochondrial protein associated with oncostatic (=Tumor inhibiting) activity. This induces mitostatin-dependent mitophagy, which leads to negative feedback regulation of vascular endothelial growth factor A (VEGF-A) production, thereby attenuating tumor angiogenesis.
Although PGC-1α expression generally counteracts the process of autophagy, it can promote mitophagy to maintain mitochondrial homeostasis in this specific scenario, further illustrating the complexity of the role of PGC-1α in regulating autophagic signaling pathways [71]. Mitochondrial biogenesis and mitophagy are tightly coupled, and a balanced interplay between these two processes is critical for cellular adaptation and stress resistance [103]. PGC-1α is a key player in these processes.
This entry is adapted from the peer-reviewed paper 10.3390/antiox12051075
This entry is offline, you can click here to edit this entry!