Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Virology
Hepatocellular carcinoma (HCC) remains a global health challenge, causing 600,000 deaths each year. Infectious factors, including hepatitis B virus (HBV), hepatitis C virus (HCV) and hepatitis D virus (HDV), have long been considered the major risk factors for the development and progression of HCC. These pathogens induce hepatocyte transformation through a variety of mechanisms, including insertional mutations caused by viral gene integration, epigenetic changes, and the induction of long-term immune dysfunction.
- hepatocellular carcinoma
- hepatitis virus
- molecular mechanism
1. Introduction
Liver cancer remains a global health challenge, with an increasing incidence worldwide. It is estimated that 1 million people will be affected by liver cancer each year by 2025 [1]. Hepatocellular carcinoma (HCC) is the most common form of liver cancer, accounting for about 90% of all cases [2]. HCC is the fifth most common cancer and the second most common cause of cancer death [3,4]. Every year, there are more than 700,000 new cases of HCC worldwide, and more than 600,000 people die of HCC, among which the incidence in males is higher than that in females [5]. HCC is mainly distributed in China, Southeast Asian countries and sub-Saharan Africa, with the highest incidence of HCC in east Asia and southeast Asia, and most cases occur in the age range of 30–60 years [5,6]. In China, liver cancer is an important factor affecting people’s health, ranking second among six major cancers (lung/bronchial, liver, stomach, esophagus, colorectal and pancreatic). A study using raw data from China’s National Mortality Surveillance System to assess mortality rates for all cancers and site-specific cancers from 2004 to 2018 showed that liver cancer was an important cause of death in people < 65 years old in China, accounting for 44.35% of all cancer deaths, and HCC accounted for the vast majority of these liver cancer cases [7].
The main risk factors for HCC include liver cirrhosis, chronic hepatitis B virus (HBV) infection, hepatitis C virus (HCV) infection, hepatitis B virus and hepatitis D virus (HDV) co-infection or overlapping infection, long-term consumption of aflatoxin-contaminated food, long-term alcohol consumption, obesity, smoking and type II diabetes mellitus [2,3,5]. Infectious factors, including hepatitis B virus (HBV), hepatitis C virus (HCV) and hepatitis D virus (HDV), play a very important role in the pathogenesis of HCC, although the incidence of HCC caused by non-infectious factors is increasing. HBV infection is the most important risk factor for HCC, accounting for about 50% of the incidence of HCC [2,8]. The risk of HCV infection leading to HCC is greatly reduced by a sustained virology response (SVR) with direct-acting antiviral agents (DAAs), but approximately 30% of HCC is due to HCV infection [2,4,5,9]. Co-infection or overlapping infection of HDV and HBV will increase the risk of HCC in patients with chronic HBV infection by two- to six-fold and accelerate the progression of HCC [5]. There are significant differences in the risk factors for HCC in different regions. In sub-Saharan Africa and most Asian countries, except Japan, HBV infection is a common factor for HCC, while HCV infection is a major infectious factor for HCC in some other regions [6,10]. Overall, the Asia–Pacific region has the highest rates of HBV and HCV infection in the world, with 74% of global liver cancer deaths occurring in Asia [11].
The pathological mechanism of HCC is a complex multi-step process. The interaction of various factors leads to malignant transformation of hepatocytes and early development of HCC. These include genetic predisposition, viral and non-viral risk factors and their interactions, the cellular microenvironment composed of various immune cell involvement, and the severity of the underlying chronic liver diseases. More than 90% of HCC cases occur in patients with chronic liver disease [12,13]. Most patients with acute HBV or HCV infection will not develop HCC, and the long-term host immune response and chronic inflammation caused by the failure of acute infection clearance may be the key determinants of the progression from infection to severe liver diseases such as cirrhosis or HCC [8]. Chronic hepatitis caused by chronic viral infection and continuous immune response leads to liver tissue damage, and further causes liver fibrosis and cirrhosis. Cirrhosis is a susceptibility factor for HCC, while advanced liver fibrosis is highly associated with HCC risk [4,14]. In the process of the development of chronic liver disease and cirrhosis of the liver, components of the immune system such as the microenvironment and the function of the virus directly under the influence of liver cells gradually accumulate a large amount of DNA damage, chromosomal instability, new blood vessels in the early generation and epigenetic change, causing liver cell transformation, which is the basis of most HCC incidence [2,15].
HBV, HCV and HDV cooperatively infect cells through various mechanisms, leading to the occurrence of HCC (Figure 1). Common mechanisms among the three viruses include (1) persistent hepatitis and immune-mediated oxidative stress damage caused by chronic viral infection; (2) intracellular oxidative stress damage induced by viral proteins; (3) the abnormal regulation of cell signaling pathways by viral proteins (such as HBx, L-HDAg, S-HDAg, HCV core, NS3 and NS5A/B) [16]. As a hepatophilic DNA virus, HBV can integrate its viral DNA into the host genome and lead to cell carcinogenesis through insertional mutagenesis or the expression of viral proteins (such as HBx) [3,16,17]. Chronic hepatitis C (CHC) infection promotes metabolic reprogramming leading to steatosis, which triggers hepatitis and stimulates the development of liver fibrosis and progression to cirrhosis, and most HCV-associated HCC is based on liver fibrosis or cirrhosis [18,19,20]. HDV as a defective virus must depend on HBV to survive. Acute HDV infection can lead to more serious liver diseases through co-infection or overlapping with HBV infection, while chronic HDV infection will increase the progression rate of liver fibrosis, accelerate the progression of patients to cirrhosis, and ultimately increase the risk of HCC [21,22].
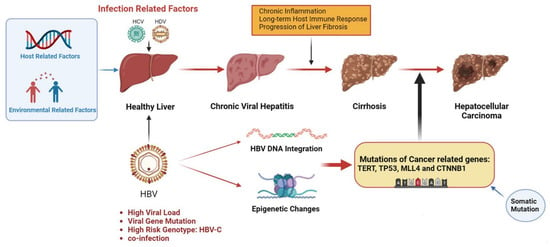
Figure 1. Pathogenesis of infection-induced hepatocellular carcinoma. Infectious factors include HBV, HCV and HDV, which mainly mediate chronic inflammation and persistent immune response through chronic viral hepatitis, leading to liver fibrosis and cirrhosis, inducing somatic mutations, and eventually leading to HCC. HBV infection is a major infectious factor with a unique oncogenic mechanism. HBV can cause cancer-related gene mutations through gene integration and epigenetic changes and promote the occurrence of HCC. Other virus-related risk factors include high viral load, viral gene mutations and high-risk genotypes. Environmental and host-related factors play a synergistic role. The figure material is referenced from Biorender.
Although the vast majority of HCC is induced by chronic liver disease, further studies have found that HBV-related HCC occurs in cirrhosis and the normal liver, indicating that viral infection may have its own unique molecular mechanism of carcinogenesis in addition to inducing chronic liver inflammation [17]. Among these molecular mechanisms, viral infection induces gene mutations in liver cells and triggers hepatocyte transformation, which is closely related to the occurrence of HCC. It has been found that approximately 25% of HCC tumors have inducible mutations. Although the prevalence of most of these mutations is low (10%), there are still some gene mutations that frequently occur in the hepatocytes of HCC patients, such as TERT, TP53, and CTNNB1. These genes are often tumor suppressor genes or proto-oncogenes in the host genome, which are closely related to the occurrence of hepatocellular carcinogenesis [23,24]. In addition to gene mutations, infectious diseases can also affect host gene expression through epigenetic regulation, leading to abnormalities in a variety of signaling pathways. Although these mechanisms driving HCC are random, some specific genetic abnormalities or epigenetic regulations are inextricably linked to HCC [25]. Thanks to the development of next-generation high-throughput sequencing technology, bio-information technology and proteomic analysis technology, we can understand the molecular mechanism of HCC from the perspectives of the genome, epigenetics, the transcriptome and proteome, and explore the gene expression patterns and epigenetic characteristics of tumor tissues and cells [3]. The discovery of novel mechanisms of viral carcinogenesis is critical, as these findings may ultimately lead to the development of vaccination strategies and HCC treatment decisions, leading to personalized medicine to reduce virus-mediated cancer mortality and improve the prognosis of HCC patients [2,26].
2. Hepatitis-B-Virus-Induced Hepatocellular Carcinoma
Hepatitis B virus (HBV) infection remains an important global public health problem with significant morbidity and mortality [26,35,36]. Despite the availability of preventive vaccines and antiviral therapy to halt disease progression and reduce the risk of liver cancer, about 257 million people worldwide are still living with chronic hepatitis B virus infection. HBV causes more than 850,000 deaths annually and is the most common cause of HCC (44% to 55%) [5,35,36]. The majority of HBV cases occur in Asia and sub-Saharan Africa, where HBV infection accounts for 60% of HCC cases, compared with only 20% in the west [36,37,38]. HCC is also an important cause of death in patients with HBV. Compared with non-cirrhotic patients, HBV-related cirrhotic patients have a 31-fold increased risk of HCC and 44-fold increased mortality [5]. Several other risk factors for HCC that increase the risk of HCC in HBV carriers include the male sex, older age, family history, viral type, and cirrhosis. In Africa, HBV-infected patients develop HCC, possibly because of exposure to aflatoxin B1, which acts synergistically with HBV and increases the risk of hepatocellular carcinogenesis [2,5].
At present, the improvement of social health conditions, hepatitis B vaccination program and effective antiviral treatment have reduced the HBV infection rate in many areas with a high prevalence of hepatitis B [39,40]. However, even with universal vaccination programs, it is not intrinsically possible to prevent acute cases of HBV infection, especially in high-risk groups. The existing antiviral treatment for HBV cannot eliminate cccDNA in liver tissue, so as to achieve a complete cure of HBV. Population movements are also currently changing the prevalence and incidence in some low-prevalence countries, as these migrants often come from areas with high HBV carrier rates [36]. In addition, the incorrect use of blood transfusions and blood products, unsafe sex, intravenous drug use and mother-to-child transmission can all contribute to human transmission of HBV.
HBV is a small enveloped hepatotropic DNA virus that replicates by reverse transcription [41]. HBV particles enter hepatocytes through the capture of liver-specific surface receptor solute carrier family 10 member 1 (SLC10A1) and Na+-taurocholate cotransporting polypeptide (NTCP) [42]. HBV contains relaxed circular double-stranded DNA (rcDNA), which is repaired to covalently closed circular DNA (cccDNA) by DNA repair enzymes after invasion of the host nucleus. cccDNA can be stably retained in the nucleus for a long time and functions as a small chromosome, which is the template for all viral mRNAs (such as pregenomic RNA (pgRNA)) and determines the persistence of HBV infection. In the cytoplasm, these viral mRNAs encode structural and regulatory proteins that are packaged into the viral capsid along with pgRNA, which is retrotranscribed with the viral polymerase to form rcDNA. Nucleocapsids containing HBV DNA are encapsulated by the host membrane, which is covered with all three forms of HBsAg, and then secreted from the cell as virions by multivesicular bodies [41,43,44]. Because of the persistence of cccDNA in the nucleus, HBV carriers are at risk of developing chronic HBV infection. With the progression of chronic HBV infection, the risk of end-stage liver disease, including cirrhosis and HCC, increases [15].
The development of HBV-induced HCC is a complex, multifactorial, and progressive process involving the interaction of the virus with endogenous mutagens and the host immune response to the virus [3,17,45]. HBV-associated HCC may also occur in non-cirrhotic livers, suggesting that in addition to stimulating host immune responses and driving chronic necrotizing inflammation in the liver, HBV also plays a direct role in liver transformation by triggering common and cause-specific oncogenic pathways [45,46]. With the development of next-generation sequencing technology and bio-information analysis technology, the molecular mechanism of HBV-induced HCC has been better understood, including key gene mutations caused by gene integration, chromosomal aberrations and epigenetic changes, and the dysregulation of cell signaling pathways caused by these mechanisms together. These signaling pathways play a crucial role in the benign-to-malignant transition of HCC [15,47]. Among these mechanisms, HBV DNA gene integration is a key step in HBV-induced HCC pathogenesis, which can regulate cellular gene expression [48]. The integration of the viral genome into the host genome can occur randomly, and although it is not necessary for viral replication, it is one of the important mechanisms of hepatocyte transformation [36]. Long-term and chronic HBV infection greatly increases the probability of viral gene integration and promotes carcinogenesis. Such integration events have been demonstrated in 75–90% of HCC tissues, and these integrations may further lead to the development of host–virus fusion transcripts [26,49]. If gene integration occurs in some key gene regions, then it may lead to the activation of some proto-oncogenes or the suppression of tumor suppressor genes. In addition, certain viral genotypes and HBV variants are also known to increase the risk of HCC. Epidemiological studies have shown that HBV genotypes C, D, and F have a higher lifetime risk of HCC development than genotypes A and B [14,36,50].
2.1. HBV DNA Integration
HBV is a small 3.2 kb DNA virus which can promote cellular transformation by genomic integration [3,17]. Although HBV uses reverse transcription for replication, unlike retroviruses, integration is not a key step in the viral life cycle and does not produce replicative viruses [51]. This integration tends to occur randomly, with partial double-stranded rcDNA formed 90% of the time during the reverse transcription of pgRNA. As the genetic material of HBV, rcDNA can be used to supplement the cccDNA library and generate live virions that can continue to infect new hepatocytes [52]. For the remaining 10% of cases, the reverse transcription process does not produce rcDNA but synthesizes double-stranded linear DNA (dslDNA). dslDNA can exist in virus particles or be integrated into host genes [52,53].
Although HBV DNA integration is random, it is closely related to the occurrence of HCC, and about 75–90% of HBV-related HCC cases have reported HBV DNA integration [14,49]. In contrast, HBV integration events are less frequently detected in adjacent non-tumor tissues [54]. In a study of 177 HBV-related HCC patients, HBV integration was found in 88% of patients at the HBV/human junction site [17]. The frequent discovery of HBV gene integration in HCC patients highlights the potential importance of this mechanism in the HBV induction of HCC. Among them, HBx and HBsAg are genes with the highest degree of integration, both of which are important for the transmission of viral replication/infection [1,55]. HBV DNA integration induces HCC mainly through three mechanisms: (1) the chromosomal instability of HBV integrated DNA; (2) changing the expression or function of proto-oncogenes and tumor suppressor genes to drive the occurrence of liver cancer; (3) the expression of integrated mutant HBV protein [52,56].
Almost half of all HCC cases are associated with hepatitis B virus (HBV) infection, and long-term chronic infection can greatly increase the probability of viral gene integration and promote carcinogenesis. Therefore, the accurate identification of HBV integration sites at the single-nucleotide resolution and understanding of the mechanism of cell carcinogenesis induced by HBV DNA integration are crucial for a better understanding of the HCC cancer genome landscape and the disease itself [26]. The application of next-generation sequencing technology and bio-information technology has advanced our understanding of HBV gene integration.
2.2. Epigenetic Changes
Epigenetic changes alter gene expression, which affects cell and tissue phenotypes. Common epigenetic modifications include DNA methylation, chromatin covalent modifications, nucleosome position changes, mRNA modifications, and changes in the levels of microRNAs and lncRNAs. Epigenetic modification and genetic changes can synergistically promote tumor occurrence, progression and metastasis [3]. HBV-related epigenetic changes regulate abnormal gene expression and affect signaling pathways, although they do not alter gene composition. These changes will promote the occurrence of cell canceration and increase the possibility of HCC.
In HBV-mediated epigenetic changes, chemical modifications of mRNA directly affect cellular transcription, translation and gene expression. Cellular RNA chemical modification can regulate RNA stability and turnover. Among several known RNA chemical modifications, n6-methyladenosine modification (m6A) is the most common internal mRNA modification in eukaryotic cells, which is usually enriched in the 3′-untranslated region (UTR) and around the stop codon of cellular mRNA [66,67]. Studies have found that HBV RNA transcripts are also modified by m6A, which has been shown to affect the life cycle and pathogenesis of HBV. However, HBV can change the expression of host genes by regulating the m6A modification of homologous RNA in host cells, so as to increase or decrease gene expression [68]. When the HBV-mediated modification of m6A affects the expression of cancer-related genes, cells become cancerous. Among the HBV-induced mRNA modifications, PTEN m6A modification is an example. PTEN is widely recognized as a tumor suppressor. PTEN is a metabolic regulator as well as a negative regulator of cell growth signaling pathways and is also involved in the regulation of innate immune responses activated by viral infection [69,70,71]. The levels of PTEN mRNA and m6A-modified PTEN mRNA in human liver biopsy specimens from healthy individuals, HBV-negative HCC patients, and HBV-positive HCC patients were compared and analyzed [72]. The results showed that HBV significantly increased the m6A modification of PTEN mRNA in cells (a two-fold increase), and m6A modification of PTEN mRNA negatively regulated its RNA and protein expression levels, led to the instability of PTEN mRNA and blocked the interferon signaling pathway. This change weakens the cancer suppressive effect of PTEN, which is conducive to the occurrence of HCC and immune escape [72]. In addition, HBV may also promote the development of HCC by activating the PI3K/AKT pathway and reducing the stability of PTEN mRNA [72].
Some members of non-coding RNAs (e.g., lncRNAs, microRNAs) are important components of epigenetic modifications and are involved in the regulation of gene expression, although they are not transcribed to form proteins. lncRNAs (long non-coding RNAs) are composed of 200–300 nucleotides and regulate gene expression through various mechanisms, including the recruitment of chromatin-modifying enzymes or the interaction with proteins to direct their binding to DNA [73,74]. Many lncRNAs have abnormal expression levels in tumor cells of HCC patients, many of which are involved in the regulation of cell adhesion, immune response and metabolism, and promote tumor progression [3,35,73,75]. The main function of the HBV HBx protein is to establish and maintain a cccDNA microchromosome with transcriptional activity. However, in addition to viral chromatin, HBx can recruit and regulate a variety of coding genes and non-coding RNA promoters [76]. In cell models related to HBV replication, researchers have found that HBx binds to the promoter region of DLEU2, enhances the transcription of DLEU2, and induces the accumulation of DLEU2 RNA in infected hepatocytes [35]. DLEU2 is a long non-coding RNA (lncRNA) expressed in the liver and increased in human HCC, which plays a role in the regulation of host target genes and HBV cccDNA [35]. This finding further demonstrated the epigenetic control of host genes by HBV and the transforming effect of HBx on liver cells in HBV-induced HCC [35]. MicroRNAs are short, consisting of 20 to 22 nucleotides resulting in non-coding RNAs that pair with the complementary 3′-untranslated region of messenger RNA, inhibiting its translation or causing its degradation. A single microRNA can control the levels of multiple messenger RNAs to regulate biological processes, such as apoptosis, differentiation and metastasis, and can play a role in tumor development [77]. MicroRNA-125a has been found to be associated with HCC development in chronic HBV infection [5,78].
By comparing the epigenetic changes in normal liver tissue cells and tumor cells of HCC patients, we can reveal the mechanisms of carcinogenesis induction, and find new diagnostic biomarkers and targeted therapeutic targets. MINPP1 is a tumor suppressor, which can inhibit tumor proliferation and metastasis. A group of researchers used microarray analysis to compare genes and microRNAs in liver tissues of HBV-positive and HBV-negative HCC patients and found that microRNA-30B-5P significantly down-regulated MINPP1 expression in hepatocytes of HBV-positive HCC patients and promoted the glycolytic bypass pathway, significantly enhancing the proliferation and migration of tumor cells [79]. However, microRNA-30B-5P/MINPP1 cannot regulate glycolytic bypass to promote tumorigenesis in HBV-negative HCC cells [79]. A further bioinformatic analysis of large cohort data showed that high MINPP1 expression was associated with better survival in HBV-positive HCC patients, which may contribute to the slower progression of the disease. These results demonstrate the potential value of miRNA-30B-5P/MINPP1 as a new biomarker for the early diagnosis of HBV-positive HCC and a potential drug target for antitumor therapy [79].
2.3. HBV Gene Mutation
HBV is currently divided into nine recognized HBV genotypes (A to I) and one to be determined (J). Genetic diversity and viral mutations cause different HBV infections to have different pathogenic characteristics and lead to different clinical outcomes. Genotype C and D infection are high risk factors for the development of HCC in patients with chronic hepatitis B [5,50]. HBV DNA sequencing results showed that HCC and cirrhosis patients with HBV integration were dominated by HBV genotype C [14]. Mutations in the HBV gene may further increase the carcinogenic risk of infection with specific genotypes of viruses. Several specific HBV variants are known to cause HBV-associated carcinogenesis, including the HBV pre-c mutation G1896A, the pre-s gene deletion, the basal core promoter (BCP) A1762T/G1764A mutation, and the HBx gene K130M + V131I + V5M double or triple mutations [45,49,50,80]. Mutations in the basal core promoter (BCP) region enhance viral replication by creating binding sites for hepatocyte nuclear factor (HNF) proteins, resulting in increased viral RNA transcription or enhanced viral encapsulation [45]. The A1762T/G1764A double mutation is the most common mutation in BCP and has been shown to significantly and independently increase the risk of HCC in patients with chronic HBV genotype C [50]. The G1896A mutation in the pre-core region (PC) can regulate HBeAg synthesis at the translational level by introducing a stop codon [81]. A specific HBV genotype F1b was found to be strongly associated with HCC in Alaska Native young adults [50]. A complete sequence analysis showed a significant increase in T1938C/A2051C mutations in the core region of HBV from this F1b genotype, including A1762T/G1764A mutations in the basal core promoter region (BCP) and G1896A mutations in the pre-core region (PC) [50]. Although the specific mechanism by which F1b genotype HBV induces HCC remains unclear, the accumulation of these core mutations can promote the development of early HCC [50].
In addition to mutations in the core region of HBV, mutations in some other regions may also be associated with the occurrence of HCC, which can be used as diagnostic and therapeutic targets for HBV-induced HCC. The HBV RT gene is a target sequence for antiviral therapy with nucleoside analogues, but due to the high replication rate of HBV, immune pressure from the host, and error-prone HBV reverse transcriptase, frequent mutations may occur, leading to the immune escape of HBV and potential carcinogenesis [82]. In a study of 307 patients with chronic hepatitis B (CHB) and 237 patients with HBV-induced HCC, researchers performed high-throughput parallel sequencing of HBV-infected RT genes in these patients and applied machine-learning (ML) algorithms to identify internal quasi-species patterns in the HBV RT region for the prediction of individual HCC. This model is effective in differentiating HCC from CHB and has impressive predictive performance [80]. Hepatitis B virus X (HBx) mutations also increase HCC. Among them, HBx combination mutants and the carboxylic-acid-terminal-truncated HBV X protein (Ct-HBx) are considered to have a higher carcinogenic risk [83,84]. HBV integration into the host genome often results in the truncation of the HBV genome, especially at the C-terminus of hepatitis B virus X (HBx), resulting in the production of Ct-HBx. The most common HBV X gene encoding Ct-HBX in HBV-HCC samples significantly increased the aggressiveness of HCC compared with the full-length X gene [84]. A team of researchers injected wild-type HBx (WT-HBx) and four HBx mutants (M1: A1762T/G1764A; M2: T1674G + T1753C + A1762T/G11764A; M3: C1653T + T1674G + A1762T/G2764A; and Ct-HBx) into mice to observe the association of HBx mutations with HCC. [83]. The results showed that the incidence of HCC was higher in mice injected with M3-HBx and Ct-HBx. By cDNA microarray analysis, M3-HBx and CtHBx significantly up-regulated the expression of plasminogen activator inhibitor-1 (PAI1) and cell division cycle 20 (CDC20) in the liver and induced pro-cancer inflammation to promote carcinogenesis [83]. Silencing PAI1 attenuates the effect of this mechanism on cells. PAI1 can be an important predictive and prognostic biomarker and a promising therapeutic target for HBV-HCC [83]. Considering the high rate of HBV gene mutation and its close association with HCC, some specific inducers of HBV gene mutation can also be used as HCC-specific biomarkers. APOBEC3G (A3G) cytidine deaminase is an innate immune limiting factor that edits and inhibits hepatitis B virus (HBV) replication. A3G induces specific HBV gene mutations through deamination, which can provide a basis for HCC screening [85].
2.4. Progress in Treatment
Early prophylaxis and treatment are the strategies of choice to reduce the incidence of HBV-induced HCC, including vaccination to prevent HBV infection, the monitoring of hepatic fibrosis progression with reliable biomarkers, and the use of effective antiviral therapies such as nucleo(s/t)ide analogue (NAs), entecavir (ETV), tenofovir alafenamide fumarate (TAF) and tenofovir disoproxil fumarate (TDF) [2,5,86,87]. These measures can greatly change the prognosis of patients with chronic HBV infection, thereby improving liver histology, preventing progression into HCC, and improving the survival rate [5]. However, vaccination does not completely prevent HBV infection. The existing antiviral therapy also has difficulty in achieving a complete cure of HBV infection, because these drugs cannot eliminate cccDNA in liver tissue [35]. Every year, many HBV-infected patients still develop CHB, which greatly increases the possibility of HCC. The conventional treatment of HCC includes surgical treatment and non-surgical treatment. Surgical treatment is the main treatment for HCC patients, including hepatectomy and liver transplantation. Chemotherapy and radiotherapy are commonly used for HCC patients who cannot be treated surgically. Radiofrequency local ablation is the main method for the non-surgical treatment of early HCC guided by imaging [88]. In terms of drug therapy, HCC is relatively resistant to traditional chemotherapy drugs, such as 5-fluorouracil, cisplatin, doxorubicin or gemcitabine [3]. The emergence of targeted molecular drugs, such as sorafenib, lenvatinib and other second-line medications, has led to better supportive care for patients with advanced HCC who cannot be operated on. Despite the remarkable clinical efficacy of molecular therapies in patients with advanced HCC, drug resistance almost inevitably emerges, hindering eventual cure. Cancer heterogeneity and the misjudgment of clonal drivers are the main reasons for the failure of molecular therapy and the induction of drug resistance [89]. Regional and ethnic differences also lead to significant heterogeneity in these treatments [6]. At present, new molecular mechanisms related to HBV-induced HCC disease have been continuously discovered, which also creates more potential for the development of new targeted therapies. Given that HBV is a virus with a reverse transcriptase replication step, some of the mechanisms used to inhibit infection with retroviruses such as HIV may also be effective. SLFN11, a member of the human Schlafen family, has previously been shown to inhibit the production of human immunodeficiency virus 1 (HIV-1) through codon usage [90]. Recent studies have found that SLFN11 expression is decreased in HCC, suggesting that SLFN11 may play a role in inhibiting HBV-induced HCC tumor progression. A further mechanism analysis revealed that SLFN11 may inhibit the mTOR signaling pathway through RPS4X, while the mTOR pathway inhibitor INK128 could achieve a similar tumor suppression effect. This study suggests that INK128 and SLFN11 can be used as new therapeutic strategies for HCC [91]. Similarly, some targeted drugs used in the treatment of other tumors may also be effective in HCC. Recent studies have found that the use of the SOAT1 inhibitor AvasimiBE can inhibit the proliferation and migration of HCC cells, and high levels of SOAT1 expression have previously been shown to be associated with a poor prognosis in prostate and pancreatic cancer [92].
The HBV-induced persistent host immune response and abnormal regulation of the immune system play a very important role in the pathogenesis of HBV-induced HCC [8]. Immunotherapy for HCC including checkpoint inhibitors and monoclonal antibodies is one of the research hotspots. Some immune checkpoint inhibitors, such as Nivolumab (anti-PD-1 mab), have been approved for the treatment of advanced HCC patients treated with sorafenib in several countries and regions, and have shown reliable safety [6]. Recent studies have demonstrated that HBV contains α2,6-biantennary sialoglycans that can regulate the host immune response by binding to sialic-acid-binding IgG-like lectins (SIGLECs), which may contribute to HBsAg-induced immunosuppression. Based on this result, researchers found that the combination of anti-SIGLEC-3 mab and GS-9620 could activate host immunity to produce anti-HBsAg antibodies, clear HBsAg, and reduce the incidence of HCC in CHB patients [93].
Compared with traditional HCC treatment methods, the use of systemic therapies, including immune checkpoint inhibitors (ICIs), tyrosine kinase inhibitors (TKIs), monoclonal antibodies and other targeted molecular drugs can better achieve personalized treatment, especially to improve the survival rate of advanced patients who cannot be treated with surgery [2]. The continuous exploration of the disease mechanism of HBV-related HCC is crucial for us to enrich therapeutic methods and deal with tumor resistance.
This entry is adapted from the peer-reviewed paper 10.3390/cancers15020533
This entry is offline, you can click here to edit this entry!