Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Materials Science, Composites
Innovation in the design of Li-ion rechargeable batteries is necessary to overcome safety concerns and meet energy demands. In this regard, a new generation of Li-ion batteries (LIBs) in the form of all-solid-state batteries (ASSBs) has been developed, attracting a great deal of attention for their high-energy density and excellent mechanical-electrochemical stability.
- all-solid-state batteries
- solid-state electrolyte
- li-ion batteries
1. Brief History of Lithium-Ion Batteries (LIBs)
Research on lithium-ion batteries (LIBs) began during the oil crisis in the 1970s, when scientists pondered options for alternative energy sources and the potential of rechargeable devices. Stanley Whittingham, a chemist at Exxon mobile, devised a novel battery design with titanium disulfide as the cathode and lithium metal as the anode, which could be charged in a short period of time (Figure 1a). It was not successful, however, due to the thermal runaway evident in early tests. An engineering professor and physicist, John B. Goodenough from the University of Texas in Austin, advanced it by swapping out the titanium disulfide cathode for lithium cobalt oxide (LCO), thereby doubling the energy capacity of the battery (Figure 1b) [11]. Five years later, Akira Yoshino from Meijo University in Nagoya, Japan, applied a carbonaceous material instead of the lithium metal anode, which proved ground-breaking, as it was the first prototype of a lithium-ion battery without lithium metal (Figure 1c). For developing this “rechargeable technology”, the three aforementioned scientists shared the Nobel Prize in 2019.
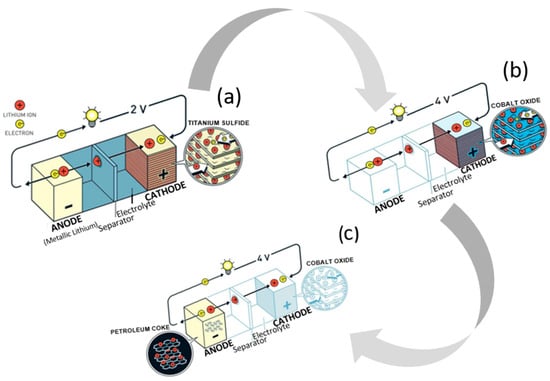
Figure 1. Battery designs by Whittingham (a), Goodenough (b), and Yoshino (c).
In 1997, a research team led by Goodenough introduced the cathode material of lithium iron phosphate (LFP), an alternative to LCO, since CO is high in toxicity and more expensive. LFP has the advantages of low material cost, nontoxicity, a 3.5 V operating voltage (in contrast with Li/Li+), a considerable theoretical specific capacity of 170 mAh g−1, high stability, and prolonged cycle life. Compared to other cathode materials, though, the electronic conductivity of the LPF cathode is limited to 10−9 to 10−10 S cm−1 [12,13]. Other drawbacks include a poor ion diffusion rate, low tap density, and unacceptable electrochemical performance at low temperature, limiting its further development [14,15,16]. A layered cathode material was proposed later, such as the combination of nickel, manganese, and cobalt (LiNi1−x−yCoxMnyO2; referred to as the NMC type of cathode), which boasts high specific capacity, low internal resistance, and heightened safety. Forms of NMC cathode with less cobalt content (e.g., NMC811, NMC442, NMC532) have garnered attention as cheaper options, and a great deal of research has been invested in developing cobalt-free alternatives. Comparing two designs capable of delivering 4.3 V, the greater amount of nickel in a cathode such as NMC811 facilitates a higher specific capacity of 200 mAh g−1 than NMC532 (160 mAh g−1), versus Li/Li+ configurations. The increased level of Ni content in the cathode raises the reactivity of the cathode, however, due to the instability of the nickel ion with the liquid organic electrolyte, resulting in two-times the extent of moisture. This is why nickel-rich cathode materials need an additional electrode coating to prevent degradation during operation at high temperature [17,18].
Besides LIBs, a step-change in energy storage is the appeal of technologies that differ from Li-ion-based systems. Lithium-air (Li-air) and lithium-sulfur (Li-S) are theoretically capable of providing the kind of the performance required for the future. Aqueous and non-aqueous Li-air batteries were first described in the literature in [19] and [20,21] respectively. The concept of electrochemical energy conversion and storage, employing sulfur as the cathode in an alkali metal anode battery date back to at least 1960 [22]. Reactions at the cathode (the positive electrode) in Li-air and Li-S cells involve the reversible reduction of O2 and S, respectively, and are fundamentally different from those in Li-ion cells. Although theoretically the energy densities of Li-air and Li-S cells are high (Table 1), numerous issues need to be addressed prior to shifting the technology from theory to practice [23].
Table 1. Theoretical energy storage for LIBs.
| Battery Chemistry | Cell Potential/V | Theoretical Specific Energy/Wh kg–1 |
|---|---|---|
| Li-S2Li + S = Li2S | 2.2 | 2567 |
| Li-air (non-aqueous) 2Li + O2 = Li2O2 | 3.0 | 3505 |
| Li-air (aqueous) 2Li + ½O2 + H2O = 2LiOH | 3.2 | 3582 |
| Contemporary Li-ion 0.5C6Li + Li0.5CoO2 = 3C + LiCoO2 | 3.8 | 387 |
A shuttle effect reported for lithium polysulfide (LiPSs) and slow sulfur reaction kinetics, caused by multi-step phase transitions, severely limit the practical application of Li–S batteries. These limitations can be overcome, though, by adopting a facile hydrothermal method and performing defect engineering to synthesize a cocklebur-like sulfur host with a TiO2-VOx heterostructure (CTVHs) in the production of long-life Li–S batteries [24]. Heterostructures have the potential to aid the development of new Li–S batteries or other energy storage systems, and could find widespread application in various interface control solutions.
2. Theoretical Aspects of Li-Ion Battery Technology
2.1. Key Parameters of LIB Development
For the large-scale applicability of LIBs, such as in EVs and a smart grid arrangement, it is necessary to consider several factors. Putting LIBs in EVs, beyond energy-related concerns, means addressing several matters including cost, cycle life, safety, and environmental impact. In a smart grid setting, the related cost, safety, and life cycle are more important than energy density [25]. Modern LIBs are limited to a gravimetric energy density of ˂250 Wh kg−1 and volumetric energy density of ˂650 Wh L−1; an increase in these is anticipated of up to ~500 Wh kg−1 and ˃1000 Wh L−1, respectively. Such performance parameters largely depend on the properties of the anode, cathode and electrolyte materials employed in the battery system, the given environment and intended use.
2.2. Solid Electrolyte Interface (SEI) Formation
Cell reactions that pose a challenge to LIB technology include the occurrence of a solid electrolyte interphase (SEI), electrolyte flammability, the dissolution of electrodes, and dendrite growth [26]. The formation of SEI in an advanced rechargeable battery system arises when such a battery is operated beyond the thermodynamic stability window of the electrolyte [27]. The interphase stems from the sacrificial decomposition of electrolytic components, such as solvent, salts, and additives, resulting in the formation of a thin film which separates the electrolyte from the electrode [28]. It has been proven that the composition of such an SEI is vital to superior performance of LIBs [29]. Nevertheless, lithium ions are consumed in the presence of an excessive SEI layer during delithiation, leading to capacity fade, a rise in impedance and creation of a barrier at the anode/electrolyte interface [30,31,32,33]. Decomposition of formed SEI also initiates a chain of reactions, and further results in thermal runaway of the LIB [30].
2.3. Safety Concerns
Another critical concern about LIBs is that of safety [34], and three main categories exist: (1) the reactivity of the material under conditions of abuse; (2) flammability of the electrolyte; and (3) the toxicity of the substance if released into the environment through a crack in the cell package [35]. Reactions inside the battery that will result in thermal runaway of the system are classified as anode–electrolyte, cathode–electrolyte, and cathode–anode reactions. Commonly used organic electrolytes have a favorable operating voltage window, yet under extreme conditions of temperature and voltage they may react with the electrodes and release a significant amount of heat and gas, effecting damage to other materials or failure of the same inside the battery [36]. Anode–electrolyte reactions are known to initiate a rise in heat production, while cathode-electrolyte and anode-cathode reactions trigger a combustion process; the latter only arising when a considerable amount of heat is produced [37,38]. Such combustibility of a carbonate electrolyte polymer in batteries, leading to thermal runaway, is instigated through mechanical or thermal stress, dendrite formation, decomposition of the electrolyte, and charging issues related to electrochemical abuse [39]. During abnormal charging conditions, such as overcharging, lithium is continuously eliminated on the cathode side, inducing breakdown of the cathode and oxygen evolution. Further effects comprise oxidation of organic solvents in the system and the intense generation of heat. Moreover, excessive deposition of lithium at the anode side initiates the formation of dendrites, while the reaction of such deposited lithium and the carbonate solvent produces a huge amount of heat and gas. Very small flash points are associated with the decomposition of common lithium salt LiPF6 and the oxidation of carbonate solvents, e.g., ethylene carbonate (EC), propylene carbonate (PC), dimethyl carbonate (DMC), ethyl methyl carbonate (EMC), diethyl carbonate (DEC), and dimethyl carbonate (DME), which can be easily triggered under a state of high voltage or temperature. Should the temperature of the battery system go up, the LiPF6 salt thermally decomposes to PF5 before the solvents decompose. This PF5 is a strong Lewis acid highly reactive with organic solvents, and such a reaction could encourage the thermal decomposition of carbonate-based solvents [40,41,42]. In addition, the unusual rise in temperature and generation of heat initiates side reactions such as breakdown of the SEI layer or destruction of the separator, which constitute causes of thermal runaway of the battery system [43,44,45]. In the case of EVs, a primary issue relates to the size of the battery, namely a decrease in the ratio between the thermal cooling area and heat generated raises the risk of a fire in the battery system [46,47].
Dendrite formation represents a major issue in LIB technology. Li metal has had real appeal as an anode material for LIBs due to its ultrahigh specific capacity of 3860 mAh g−1, in addition to its low negative redox potential of −3.04 V in comparison with a standard hydrogen electrode (SHE) [48]. The appearance of lithium dendrite in Li metal batteries is usually associated with abnormal operation conditions like overcharging or charging in low temperatures [49]. Typically, lithium dendrite growth occurs in the presence of additional lithium ions that accumulate or are deposited on the anode surface, instead of being absorbed or incorporated into the anode [50,51]. As a consequence, lithium ions permeate the separator, giving rise to short circuits, safety issues, and battery failure. In addition, lithium dendrites react with the electrolyte, causing it to decompose through the loss of lithium, thereby diminishing battery capacity [52].
2.4. Cathode Materials Applicable in Lithium-Ion Batteries
Among the components in a Li-ion cell, it is the cathodes that restrict energy density and dictate the cost per kilowatt–hour. Modern cathode materials are transition metal oxides, and three classifications of them exist according to their crystal structure: (a) layered LiMO2 (M: Ti, V, Cr, Co, Ni), with a two-dimensional layered crystal structure of LiCoO2; (b) spinel oxides (e.g., LiMn2O4, LiTi2O4, LiNiO2) of three-dimensional morphology; and (c) the one-dimensional morphology of polyanion oxides such as Li2(MoO4)3, Li2Fe2(WO4), and LiFePO4. In such intercalation cathode materials, Li+ is the guest ion that facilitates ion diffusion. Layered and spinel oxides have a close-packed structure with high density, hence they possess sufficient electronic conductivity (10−1–10−4 S cm−1). Although polyanion oxides exhibit low density and poor electronic conductivity, polyanion class cathodes afford high thermal stability and greater safety than layered and spinel oxide cathodes. Figure 2 details the crystal structure of the cathode materials and their voltage profiles [53,54].
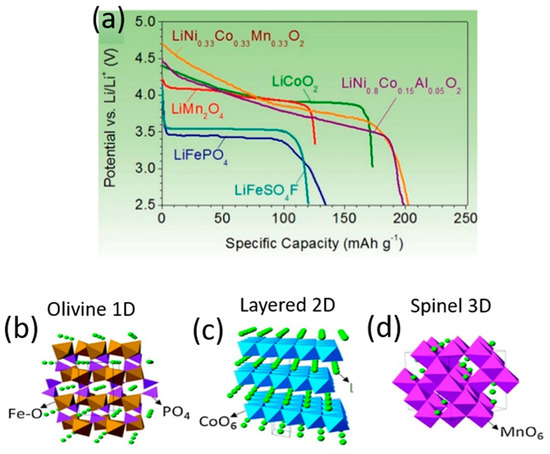
Figure 2. Discharge profiles (a) and crystal structures (b–d) of representative intercalation cathodes: olivine LiFePO4 (b), layered LiCoO2 (c) and spinel LiMn2O4 (d).
Out of the three classes of oxide cathodes, layered oxides are the preferred option. LiNi0.8Co15Al0.05 O 2 and LiNi0.33Co0.33Mn0.33O2 demonstrate the highest discharge capacity (200 mAh g−1) and are commercially applied in Panasonic batteries for Tesla EVs. However, these composite cathode materials demonstrate average discharge voltage decreases during cycling. One way of overcoming this problem is to perform surface stabilization, which minimizes volume changes, cracking, and surface reactivity [54]. It either involves applying a surface coating by chemical vapor deposition and atomic layer deposition techniques [55], or adding inactive dopant cations into the layered oxide structure. These dopants can substitute Li or transition metal cations; for example, 1–5 mol. % of Mg+2 cations lead to enhanced cycling stability. Another way pertains to the design of gradient and core-shell cathode particles. Core-shell materials are usually synthesized such that the unstable component is in the core and the thermally stable component constitutes the shell: core (Ni-rich or Li-rich) and shell (Mn-rich) [56]. For instance, the double-shelled material Li[Ni0.8Co0.1Mn0.1)2/7] core [(Ni1/3. Co1/3Mn1/3)3/14] shell-1 [Ni0.4Co0.2Mn0.4)1/2O2) shell-2 contributes to the cycling stability of the hybrid structure, resulting in superior electrochemical performance in comparison with a homogeneous cathode with the same overall composition [57]. Different engineering techniques have also been employed to increase the electrochemical performance of a cathode by removing the binder [58] and variously synthesizing carbon-based composite cathodes, e.g., by mixing in CNT and graphene oxide or by applying a coating of conducting polymers, e.g., PANI [59,60].
2.5. Anode Materials for Lithium-Ion Batteries
-
Lithium-based anodes
In order to achieve a high-energy density and fast charge capability for LIBs, it is necessary to accelerate electrochemical reactions through charge transfer at the interface. Regardless of the fact that Li metal as an anode material has a high theoretical specific capacity (ca 3860 mAh g−1) and the most negative potential (−3.040 V vs. SHE), it suffers from Li-dendrite formation, poor interfacial contact, notable volume changes and sensitivity of the electrolytes [61]. Various methods exist to regulate Li plating/striping processes, resulting in formation of “dead Li”, i.e., a protective coating or the design of a composite lithium anode [61]. Creation of a composite lithium anode requires that an additional component is introduced that possesses a similar delithiation potential, and certain reversible storage/release mechanisms of Li ions are in place to facilitate the delithiation mechanism, for example graphene. The cycling performance of a coin cell with a Li metal-graphene anode/LiFSI, DMC, HFE electrolyte/NCM523 cathode maintains 210 cycles with a capacity retention of 80%, in comparison with 110 cycles by a bare Li anode [62]. In practice, though, graphite or graphene is widely applied as an anode material in lithium-ion batteries due to the resultant favorable price-performance ratio [62].
-
Graphene-based anodes
The use of graphene as an anode in LIBs makes sense since graphene can accelerate electronic transfer and reduce contact resistance through good contact between the active materials and current collectors, as well as the electrolyte, which reduces polarization. Graphene tends to agglomerate, however, owing to π–π interaction and van der Waal forces between layers, potentially hindering its conductivity. Nevertheless, it is possible to take advantage of this property of graphene and modify its structure. In terms of structural form, graphene is available as a material in 1D (fibers), 2D (film and paper), and 3D (hydrogel forms and honeycomb-like structures). Applying such materials as anodes gives rise to superior rates (Table 2) [63].
Table 2. Performance of LIBs with graphene-based anode materials.
| Material | Amount of Graphene | Performance |
|---|---|---|
| Li4Ti5/holey-graphene | 50 wt.% | 98 mAh cm3 at 17.5 A g−1; 84% capacity retention after 1000 cycles at 7 A g−1 |
| Li4Ti5/graphene | 5 wt.% | 122 mAh cm3 at 30°C; 124.5 mA g−1; 98% capacity retention after 300 cycles at 20°C |
| Graphene-MnO2-GNRs | 68 wt.% | 300 mAh cm3 at 612 mAg−1 after 250 cycles at 0.4 A g−1 |
| MoS2-graphene | 4.7 wt.% | 570 mAh cm3 at 1A g−1; 894.1 mAh g−1 after 100 cycles at 0.1 A g−1 |
| Graphene anchored with Co3O4 | 24.6 wt.% | 484 mAh g−1 at 0.5A g−1; 935 mAh g−1 after 30 cycles at 0.1 A g−1 and a specific current of 0.05 Ag−1 |
-
Graphite-based anodes
Graphite enables enhanced full cell energy density through its low delithiation potential (0.2 V vs Li/Li+) and theoretically high gravimetric capacity (372 mAh g−1) [64].
Graphite particles are characterized by a flake-like particle morphology with two different surfaces, basal and edge planes (Figure 3) [64]. This 2D-layered structure of graphite causes the anisotropy of surface energy and influences electronic, physicochemical and mechanical properties. Weak van der Waals forces between the graphite layers enable the intercalation of ionic and molecular species across the surfaces. As a result, expansion affects the interlayer distance and re-staking of the graphite layers. A large interlayer separation is favorable for electrode materials as it facilitates lithium-ion intercalation and de-intercalation during charging and discharging. This process ends with the formation of graphite intercalation compounds (GICs), typically LiC6. GICs possess high reactivity and sensitivity to oxygen and moisture, resulting in rapid material degradation.
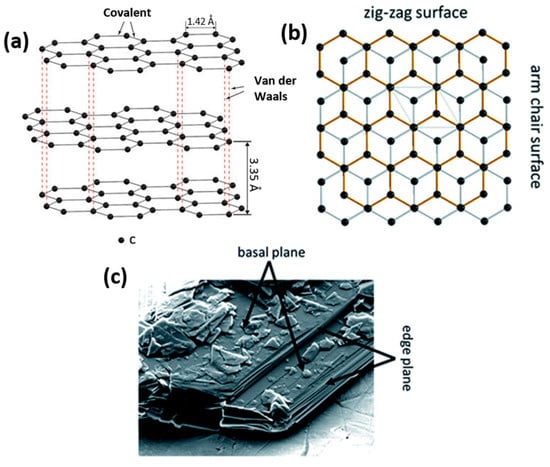
Figure 3. Schematic illustration of the layered graphite structure and the resulting presence of basal and edge planes (a) showing the difference between zig-zag and arm chair surfaces (b) and SEM micrograph of the basal and edge planes for a graphite particle (c).
-
Ti-based oxides anode materials
This category includes TiO2, Li4Ti5O2, Li2MTi3O8, MLi2Ti6O14 and others, which demonstrate excellent intrinsic safety for their high working potential (1.2–1.7 V vs Li+/Li), stable crystal structure during Li+ intercalation/de-intercalation, general abundance and low cost, but suffer from poor electronic conductivity (10−13 S cm−1) due to the highest valence state of Ti+4, thus restricting their rate capabilities [65,66].
The structural stability, pore size and specific surface area of Li2ZnTi3O8 (LZTO) co-doped with Mo6+ and P5+ ions (LZM7TP3O) can be improved by a one-step solid-state technique. When LZM7TP3O is used as the anode in a LiNi0.5Mn1.5O4/LZM7TP3O full cell, the discharge specific capacity of the full cell reaches 214.3 mAh g−1 at 0.5 C across a voltage range of 2–4.55 V for the 1st cycle [67].
Li4Ti5O2 is one is the most widely studied complex Ti-based oxides since it is easy to fabricate and boasts a stable voltage plateau, safe performance and long cycling stability; it operates in the potential window of 1.0–3.0 V, delivering a theoretical capacity of 175 mAh g−1 [68].
The crystal structure of Li4Ti5O2 possesses a spinel configuration with an Fd3m space group (Figure 4). The 3D structure of Li4Ti5O2 secures the presence of the Li-ion transport pathway, which in turn guarantees a stable voltage plateau during lithiation, preventing the formation of lithium dendrites.
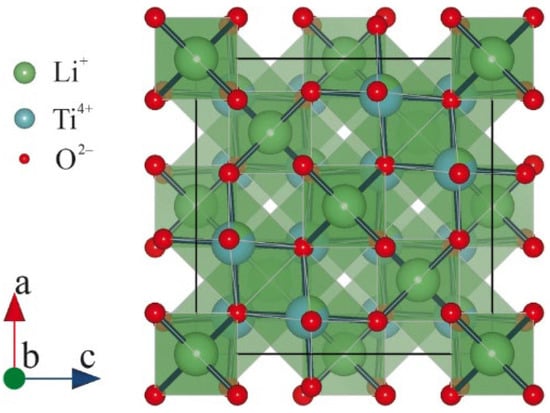
Figure 4. Crystal structures of Li4Ti5O2.
In order to enhance the rate capability of Li4Ti5O2, its composites with carbon-based materials have been fabricated by different methods, for example by mixing in CNT or by applying a coating of conducting polymers, e.g., PANI [68,69].
-
Silicon-based anode materials
At the outset, graphite-based anodes were successfully adopted to prevent dendrite formation during continuous charge/discharge cycles, and widely deployed in conventional lithium-ion batteries. However, this intercalation type of anode failed to protect the battery from dendrite growth at fast charging rates, while it also has a limited capacity, resulting in poor energy density and restricting the range of EVs. The intercalation speed of lithium-ion into a graphite structure also influences the power of the given cells. Graphite hardly meets the expectations of next generation lithium-ion batteries as a consequence. Silicon was considered as a replacement for graphite anodes, as it boasted much greater lithium storage (approx. 4000 mAh g−1). However, chemical bonds were observed to form during lithium intercalation, giving rise to a new molecular structure, in addition to which the silicon experienced swelling and contraction during a continuous charge-discharge cycle, leading to cracking and pulverization. The SEI layer deformed numerous times when cycling as a consequence, and the related side reaction consumed the lithium in the battery, causing a loss in capacity and increase in cell resistance [70]. Thus, silicon-based LIBs rapidly lose energy storage capability while cycling, and the high cost of silicon limits its applicability in large-scale usage. Numerous strategies have been developed to overcome these defects, notably application of a carbon coating, alloying and construction of porous structures [71,72]. Of the various synthetic methodologies to be researched, the deposition of Si-metal alloys shows promise as a practical means of mass-producing porous Si microparticles for the reasons of simplicity and low cost [73]. A porous Si anode prepared by dealloying Sr-modified Al–Si alloys is expected to be more effective at mitigating expansion via the introduction of abundant nanoparticles [74]. Compared to graphite and silicon anode-based LIBs, lithium metal may still be the best candidate for several reasons; although the associated drawback of dendrite formation, which diminishes the safety and service life of a lithium metal-based LIB, is likely to restrict its utilization in future high-energy devices.
In addition to the anodes suitable for LIBs mentioned above, an alloying type such as aluminum anodes show potential since they boast a high theoretical capacity (almost 1Ah g−1) [75]. Their disadvantage lies in charge transport, as the volume fraction of the surface oxide layer is significant smaller for aluminum particles (<10 μm), having the effect of severely blocking the transport of electrons. Moreover, the poor electrolyte wettability of the surface oxide layer (arising through the low affinity of the solvent molecules for the oxide layer) reduces charge transport. Grafting polar amino groups has been demonstrated as an effective means of improving electrolyte wettability [76].
3. Challenges Associated with All-Solid-State Batteries
Issues connected with LIB technology in terms of liquid electrolytes and growing demand for energy storage devices has prompted researchers to seek out alternative solutions, ushering in the era of solid-state electrolytes (SSEs) and all-solid-state batteries (ASSBs). SSEs are considered one of the best approaches for solving the lithium dendrite formation in batteries. Replacing volatile, flammable liquid electrolytes with SSEs effectively creates an impenetrable solid barrier to lithium dendrites, allowing the use of a metal lithium anode [77,78,79]. In an ideal situation, the redox of Li-ion is the only reaction to occur at the anode side, and the lithium stripping and plating are supposed to be homogeneous in ASSBs. It has proven difficult to fabricate such superior ASSBs, though. This is due to the presence of side interfacial reactions that cause instability and the formation of dendrites at the interface of the lithium metal anode, as well as low ionic conductivity and poor physical contact at the cathode interface. This subsequently leads to interfacial degradation, poor cyclic stability, reduced operating speed, and space charge formation layers, among other issues [80].
Interfacial issues that pose challenges in the manufacture and scaling up of ASSBs mainly arise from the instability of SSE. The chemical potential of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of the electrolyte determines the stability of the electrolyte. Thus, an interface is thermodynamically stable if the chemical potential of anode and cathode materials is situated between the LUMO and HOMO. Otherwise, an interlayer can appear at both the anode and cathode interface if the chemical potential of the Li metal anode exceeds that of the LUMO. and chemical potential of cathode is less than for the HOMO [81]. Three types of ASSB interface exist: (a) thermodynamically stable (no chemical reactions); (b) non-passivated mixed-conductive interphase; and (c) passivated kinetically stable interphase, created by chemical reactions at the interface of the electrolyte and electrode [82,83]. The mixed-conductive interphase shows high electronic and ionic conductivity, hence decomposition of the electrolyte at this interphase is spontaneous and promotes reduction in the electrolyte [84,85]. However, even in the kinetically stable interphase, reduction in SSE is spontaneous. Nevertheless, the interphase is electronically insulated and the electronic barrier potential decreases across the interphase [86,87]. The chemical reaction at this interface results in a stable solid electrolyte interface with diminished electronic conductivity, further limiting the possibility of additional side reactions. The formation of thermodynamically and kinetically stable interfaces could prove beneficial for the long-term performance of the battery. However, the majority of the reported solid electrolytes are thermodynamically unstable as regards the lithium metal anode, and a kinetically stable, mixed-conductive interface is often observed on the anode side of the solid electrolyte [88,89].
A major problem affecting a solid-state electrolyte is low ionic conductivity. The ion conduction mechanism in SSEs is totally different from that of liquid electrolytes. In the liquid environment, solvated ions involved in the solvation process move easily through the solvent medium [90]. In SSEs, diffusion by the mobile ions through the nonuniform environment is needed to overcome several energy barriers, including motional energy, interactions with the inherent lattice, and electrostatic forces, whereby they greatly influence the ionic conductivity [91,92]. Ion mobility in a crystalline solid also depends on interstitial aspects and interstices, among other things. Crystalline solid materials are composed of coordination polyhedral and spatial arrangements of several mobile species. The polyhedral framework enables numerous vacancies to be distributed within itself. The hooping of the mobile ion through these vacancies contributes to ion transportation. The interface between the cathode and the solid electrolyte is thermodynamically unstable due to unavoidable chemical reactivity at the interface and the restricted electrochemical potential window of the electrolyte [93,94]. At high voltage, the electrolyte tends to decompose, effecting reduction in ionic conductivity. The formation of a space charge layer (SCL) at the solid-solid interface, triggered by a substantial difference in the electrochemical potential of the cathode and the solid-state electrolyte, is another matter for consideration in the rational design of ASSBs. The SCL formed induces a Li-depletion layer, which hinders the transportation of lithium at the interface, especially in sulfide electrolytes [95,96,97,98].
Solid-State Electrolytes
Studies on SSEs have long been published, and yet huge challenges to their implementation still persist. In order to supersede an organic liquid electrolyte and avoid the safety limits of the existing LIBs, the proposed solid-state electrolytes have to satisfy the following criteria [99,100,101]:
-
− Good ionic conductivity and negligible electronic conductivity with a wide working temperature range.
-
− A chemical potential range between that of the Li metal anode and the corresponding cathode.
-
− Negligible grain boundary resistance and interface resistance at the electrode-electrolyte interface.
-
− For high temperature operation, thermal and mechanical properties, e.g., the thermal expansion coefficient, match those of the anode and cathode.
-
− High chemical stability, in connection with that of the metal anode and high voltage cathode.
-
− Low cost, environmental safety, easy to scale up and prepare.
SSEs tend to fall under three categories-solid inorganic electrolyte (SIEs), solid polymer electrolyte (SPEs), and solid hybrid electrolytes (SHEs).
-
Solid inorganic electrolytes
A solid inorganic electrolyte normally possesses high ionic conductivity, a wide stability window greater than 4.0 V, and excellent thermal stability over 100℃ [80]. Several drawbacks are associated with SIEs, however, as follows:
-
− High fragility.
-
− Poor contact with the electrode and inferior interfacial charge transport leads to high impedance.
-
− Dendrite growth and propagation through grain boundaries, especially at lower current densities
-
− High cost and poor environmental stability.
In the case of SIEs, high ionic conductivity is achieved through the transfer of ions through voids, layered structures, and excess metal ions. The observed ionic conductivity in the SIEs is in the order of mS cm−1(see Table 3). High ionic conductivity is achieved in SIEs by reducing the electrostatic force within the material, which enables the transfer of multivalent cations. Reduction in electrostatic force can be accomplished by increasing the distance between the mobile cation and the nearby anions through the anion framework by ion substitution.
Table 3. Ionic conductivity of solid inorganic electrolytes at room temperature.
| Solid-State Electrolyte | Composition | Ionic Conductivity S cm−1 |
|---|---|---|
| NASICON-like | Li1.4Al0.4Ti1.6[PO4]3 | 1.12·10−3 |
| LISICON-like | Li3.25Ge0.25P0.75S4 | 10−2 |
| thio-LISICON | Li9.54Si1.74P1.74S11.7Cl0.3 | 1.25·10−1 |
| Li3.25Ge0.25P0.75S4 | 10−2·10−3 | |
| Li9.54Si1.74P1.44S11.7Cl0.3 | 2.5·10−2 | |
| Anti-perovskites | Li3OX (X = Cl or Cl or Br) | >10−3 |
| Garnets | Li6.55La2.5BaZrTaO12 | 6·10−3 |
| Li6.5La3Zr1.5Ta0.5O12 | 0.75·10−3 | |
| Li6.25La3Zr2Al0.25O12 | 0.68·10−3 | |
| Li6.25La3Zr2Ta0.25Ga0.2O12 | 1.04·10−3 | |
| Li2S-SiS2 based | 95[0.6Li2S0.4SiS2]. 5Li4SiO4 | 10−3 |
| Li2S-P2S5 based | 70Li2S-30P2S5 glass ceramic | 3.2·10−3 |
-
Solid Polymer Electrolytes
Compared to SIEs, SPEs boast numerous advantages, including low cost, ease of fabrication, chemical and thermal stability, the capacity for large-scale manufacture, and mechanical toughness above the glass transition temperature. The strong adhesion of SPEs means they are highly compatible with the electrode material. However, the applicability of SPEs in batteries is also hindered by certain matters. Firstly, the ionic conductivity of most SPEs is limited to between 10−6 and 10−5 S cm−1, which affects the overall resistance of the battery system [102]. In an ideal case, the lithium migration number is one. If Li+ migration is too little, then the anions accumulate on the electrode surface and cause concentration polarization. Two methods are adopted to avoid this in SPEs, the first involves grifting anions into the polymer backbone (a common method for preparing single ion conductors), while the other adds an anion acceptor into the polymer matrix to restrict anion movement.
Owing to the merits of SIEs and SPEs, the development of a composite that combines the best of both is of particular interest, and these are referred to as hybrid solid electrolytes or composite electrolytes.
-
Solid hybrid electrolytes
As mentioned above, both types of solid electrolyte possess disadvantages. To overcome these issues researchers devised a composite solid-state electrolyte with a combination of polymeric and ceramic structures called a solid hybrid electrolyte (SHE). Through a proper management of composition and an appropriate fabrication process, a resulting polymer-ceramic composite electrolyte could be synthesized with desired and anticipated properties; these include acceptable room temperature Li+ conductivity, ideal mechanical strength, an extended electrochemical stability window, an improved Li+ transference number, favorable interfacial contact with electrodes, and dendrite-suppression capability [103]. A polymer matrix is needed to create a SHE, which is filled with an inorganic ceramic filler to increase the ionic conductivity of the composite material. Two groups of such inorganic ceramic fillers exist-passive and active [104]. Passive fillers do not exhibit ionic conductivity themselves, but can promote the ionic properties of polymer matrices. Passive fillers that have been utilized in the studies of Li+-ion composite electrolytes include Al2O3 [105], LiAlO2 [106], TiO2 [107], SiO2 [108], Y2O3 [109], ZrO2 [110], and Mg2B2O5 [99]. Active fillers comprise Li-ion conducting materials such as LISICON-type [111], NASICON-type [112], garnet-type [113], perovskite-type [114], sulfide electrolyte [115], and oxy-nitride electrolyte [116].
This entry is adapted from the peer-reviewed paper 10.3390/batteries9030157
This entry is offline, you can click here to edit this entry!