Cancer stem cells (CSCs) have been identified and characterized in both hematopoietic and solid tumors. Many studies showed that CSCs can be identified and isolated by their expression of specific cell markers, such as ALDH, Nanog, Sox2, OCT3/4. The significance of CSCs with respect to tumor biology and anti-cancer treatment lies in their ability to maintain quiescence with very slow proliferation, indefinite self-renewal, differentiation, and trans-differentiation such as epithelial–mesenchymal transition (EMT) and its reverse process mesenchymal–epithelial transition (MET). The ability for detachment, migration, extra- and intravasation, invasion and thereby of completing all necessary steps of the metastatic cascade highlights their significance for metastasis. In addition, CSCs comprise the cancer cell populations responsible for tumor growth and cancer metastasis, resistance to anticancer therapies.
1. Theory of CSCs and Their Characteristics
1.1. The Development of the CSC Theory and Their Discovery
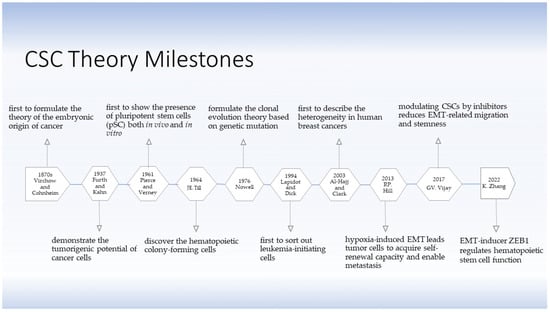
The theory of the existence of stem cells (SCs) and CSCs was developed several decades ago. The CSC hypothesis applies the concept of SCs derived from embryogenesis to understand tumorigenic processes. As early as in the 1870s, Rudolph Virchow and Julius Cohnheim were the first to propose a theory on the embryonic-like origin of cancer, linking the similarities between neoplasms and embryonic tissue by what is known as CSCs now [
4] (
Figure 1). The theory did not attract much attention until 1937, when Furth and his colleagues intravenously inoculated small numbers of leukemic cells into mice in a limiting dilution experiment and found that a single malignant leukocyte was capable of producing leukemia, thereby describing the tumorigenic potential of certain tumor cells [
5]. In 1961, Pierce and Verney discovered the multi-potentiality of ‘embryonal carcinoma cells’ from teratocarcinoma in both in vivo and in vitro experiments [
6]. They demonstrated in 1964 that these ‘embryonal carcinoma cells’ were heterogeneous in their capabilities, giving strong support to the CSC theory [
7]. In the same year, Till and his colleagues discovered that there were relative few hematopoietic ‘colony-forming cells’ present in hematopoietic tissue with capacity of extensive proliferation, differentiation and self-renewal [
8]. Nowell stated the clonal evolution theory based on acquired genetic lability in 1976 [
9], postulating that acquired genetic susceptibility allows sequential selection of variant sublines which are increasingly abnormal, explaining a stepwise tumor progression. In addition to hematologic malignancies [
10], CSC-related investigations on solid tumors, including melanoma [
11,
12], breast cancer [
13], colon cancer [
14], and lung cancer [
15], were performed. Benefitting from rapidly developing technologies, such as fluorescence-activated cell sorting (FACS) and magnetic-activated cell sorting (MACS), scientists continued to study more about CSCs. In 1994, Lapidot and Dick first sorted out leukemia-initiating cells based on the expression of cell surface markers, i.e., CD34
+CD38
− cells, estimating that approximately one in a million leukemia cells were capable of tumor initiation [
10]. In consistency, Al-Hajj and Clarke found that a minor proportion of breast cancer cells isolated as CD44
+CD24
−/low lineage had the ability to form sustainable neoplasia, and based on this difference in cell surface markers, they distinguished the tumorigenic cancer cells from non-tumorigenic ones, describing for the first time the heterogeneity of human breast cancers in 2003 [
13]. Taken together, these studies confirmed the existence of CSCs, consolidated the role and significance of CSCs in tumor initiation, development, growth, and recurrence, and supported the theory of CSCs concretely and reliably.
Figure 1. The development of the CSC theory. The theory of SCs and CSCs was initially conceived one and a half centuries ago. Virchow and Cohnheim first proposed the embryonic origin of cancer in 1870s. It took more than half a century until Furth and Kahn successfully established a mouse leukemia model indicating the possibility of tumor initiation by a minority of cancer cells. Since then, the CSC theory has been under challenge until more knowledge was accumulated. Today, the theory of CSCsthe has been substantiated and is regarded as reliable. CSCs have been demonstrated to be closely associated with EMT and metastasis [
4,
5,
6,
8,
9,
10,
13,
16,
17,
18].
In 2013, Hill and his colleagues proposed that under hypoxia condition within the tumor microenvironment (TME), disseminated cancer cells acquired self-renewal capability during an EMT-enabled metastasis process. EMT induced loss of epithelial cell polarity and loss of epithelial markers, resulting in increased cell motility and promoting the maintenance of stemness [
16]. In 2017, Vijay and her colleagues demonstrated that inhibition of glycogen synthase kinase β (GSK3β) decreased mesenchymal properties and downregulated the EMT-related migration. Further, inhibition of GSK3β significantly repressed the sphere formation, reduced stem cell properties and selectively killed mesenchymal cancer cells but not those with epithelial properties [
17]. Zhu et al. demonstrated the EMT-inducer Zeb1 regulated the function of hematopoietic stem cells (HSCs) by suppressing the mitochondrial fusion [
18]. Those supportive studies revealed the complex association of CSCs, EMT and metastasis.
1.2. The Characteristics of CSCs
Over the past decades, the existence of CSCs has been demonstrated in either hematological malignancies or solid tumors, including leukemia [
10,
19,
20]
, breast cancer [
13,
21,
22,
23], bone cancer [
24], brain cancers [
25,
26], colon cancer [
14,
27], head and neck squamous cell carcinoma (HNSCC) [
28,
29], lung cancer [
15], liver cancer [
30,
31], melanoma [
11], and ovarian cancer [
32,
33]. While most of the ground laying work on CSCs was performed in breast cancers, the principles are transferrable to other cancer types as well and are basic principles that can be applied in tumor biology of other solid tumors. In general, CSCs in different malignancies share certain characteristics.
1.2.1. Self-Renewal and Aberrant Differentiation
Both normal SCs and CSCs possess self-renewal and differentiation capacities [
34]. Generally, CSCs undergo asymmetric division with unequal cell size and distinct cell fates, i.e., one CSC generates a daughter CSC with unlimited proliferative potential through self-renewal, while it also generates a phenotypically distinct cancer cell with a limited lifespan and proliferative capacity which integrates into the tumor mass through differentiation-like mechanisms [
35]. There have been different in vitro clonogenic assays, including sphere formation, organoid culture, and co-culture assays, rapidly developed to investigate the tumor heterogeneity and to address the potential of proliferation, differentiation, and self-renewal of CSCs.
In earlier studies, a sphere-formation assay was established to study the neuronal stem cells from adult brain tissue and demonstrate the capacity of forming free-floating spheres in serum-free culture medium, called neurospheres [
36]. Coherently, Wei et al. reported a population of human skeletal muscle cells that maintained their proliferative capacity and those cells expanded in vitro as myospheres for more than 20 passages, also termed myosphere-derived progenitor cells (MDPCs). The MDPCs expressed skeletal progenitor cell markers Pax7, ALDH1, Myod and Desmin, as well as stem cell markers Nanog, sex determining region Y-box2 (Sox2) and Oct3/4 [
37]. Further, these cells were capable of spontaneous differentiation into myotubes and other mesodermal cell lineages, indicating the self-renewal and differentiation potential of SCs [
37]. By investigating different tumor entities, we showed that those stem cell markers are shared by CSCs of different cancers [
22,
28,
33]. By comparing a 2D-monolayer cell culture under normal adhesion and serum-containing culture conditions with a 3D-sphere culture under low-adherent and serum-free culture conditions, we observed that breast cancer stem cells (BCSCs) were enriched by growing out and forming spheroids in vitro with a higher enzyme activity of aldehyde dehydrogenase (ALDH) [
22]. Additionally, in previous studies of HNSCC [
28], oropharyngeal squamous cell carcinoma (OSCC) [
38], and ovarian cancer [
32,
33], we demonstrated consistently that ALDH was a reliable marker for CSC identification. The ALDH
+/high cells were capable of indefinite self-renewal and tumor initiation by xenografting into animal models [
39]. From our group, Xu et al. investigated the expression of the CSC marker ALDH1A1 in clinical specimen of HNSCC and OSCC. She found that ALDH1A1 was associated with poorly differentiated tumor histology independent of the patients’ HPV and smoking status, and patients with a higher frequency of ALDH1A1-expressing cells turned out to have a significantly shorter overall survival (OS) than those with a lower frequency. She also characterized the co-expression of ALDH1A1 and Twist1 in primary tumor and metastatic lymph node; however, no correlation was observed [
40].
In addition, many cell markers, such as the epithelial cell-adhesion molecular (EpCAM), ESA, Nestin, disialoganglioside2 (GD2), CD47, CD49f, CD61, CD117, CD133, CD166, chemokine-activated G protein-coupled receptor4 (CXCR4) and its ligands CXCL12, and ATP-binding cassette-transporters G2 (ABCG2), are commonly expressed and shared as CSC markers by different solid tumor types [
41]. Moreover, the well-established stemness conferring transcription factors (TFs) Nanog, Oct3/4, and Sox2 are applied in the CSC marker catalog, as they are involved in regulating stemness properties [
34,
41,
42]. To further characterize the CSCs, additional protein markers associated with stemness and EMT have been reported, such as the overexpression of Sox9 and Snail in BCSCs [
43,
44,
45].
1.2.2. Quiescence/Dormancy
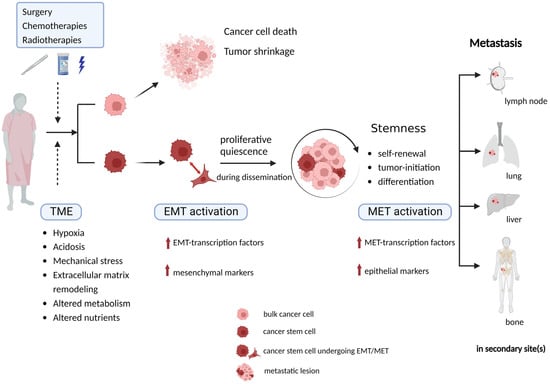
Targeting highly proliferative cells at different cell cycle phases, traditional chemo/radiotherapies induce DNA damage and trigger cell apoptosis. While CSCs can proliferate at a relatively rapid rate to repopulate the progeny pool in a fast-growing tumor, they can also switch into a quiescent state [
46]. They have the ability to stay dormant and undivided for long periods until there are favorable conditions to re-stimulate their proliferation; therefore, during EMT and metastasis, CSCs display slow cycling features during dissemination [
46,
47] and can re-enter rapid proliferation in a new TME [
35,
48]. Quiescence, also termed dormancy occurs when cancer cells are alive, but their proliferation stops. It is a reversible status because quiescent CSCs can retain the capacity to return to a proliferative state. This indicates that within an invasion–metastasis cascade, CSCs will sustain in a quiescent phase during migration, trafficking through blood and/or lymph vessels and invasion into target site(s) and when adapting to a new TME in a distant organ [
49]. It can explain the delayed recurrence of a tumor at the primary site or somewhere else in the body years after the first diagnosis [
49,
50].
Quiescence is an important feature for CSC-related resistance to adjuvant therapies, under which CSCs remain arrested in G0/G1 phase of the cell cycle to survive and to undergo cellular reprogramming to adapt to the TME [
1,
46]. Moreover, the expression of some significant CSC markers and TFs, such as CD34, Nanog, Sox2, and Oct4 (also known as POU5F1), have only been reported to be elevated in some dormant cancer cells [
46,
51]. Benefiting from the flexible state-switching ability, CSCs are highly resistant to traditional chemotherapies and radiation therapies. Consequently, CSCs retain the proliferative capacity, metastatic potential and facilitate neoplastic regrowth and progression through repeated cycles of chemo-/radiotherapy [
49]. In several solid tumors, such as colorectal cancer (CRC), pancreatic cancer, melanoma, and glioblastoma, a partial overlap has been identified between CSCs and the quiescent slow cycling cells [
48]. As detailed above, CSCs can be resistant against standard anti-tumor therapies or under circumstances of, for example, hypoxia, by maintaining a quiescent state with minimal energy consumption and slow cell cycling [
52]. In short, owing to their unique characteristics, CSCs play a crucial role in the metastatic context (
Figure 2).
Figure 2. CSCs play a crucial role in metastasis due to their unique characteristics. Undergoing conventional standard anti-cancer therapies and the multi-directional influences from TME are cellular stress factors that CSCs are supposed to survive by activating an EMT program and stay in a quiescent state to keep themselves alive during dissemination and adjustment to different conditions in a new TME, being stimulated to proliferate when successfully adjusted to distant organs/sites. TME, tumor microenvironment; CSC, cancer stem cell; EMT, epithelial mesenchymal transition; MET, mesenchymal epithelial transition.
2. Therapeutic Implications for Targeting at CSC
The existence of CSCs within a tumor provides an explanation for clinical treatment failures. CSCs can be stimulated by the therapeutic pressure, as well as changes and/or stimuli within the TME, and display a high phenotypic plasticity, accordingly, with distinct functional appearances [
93]. CSCs are closely involved in tumor initiation, development, metastasis and recurrence due to their intrinsic stemness properties and their ability to undergo the EMT process. It is reasonable to regard CSCs as a potential therapeutic target to prevent metastasis and to revert the chemo-/radioresistence. Yang et al. showed that ALDH
+ BCSCs were responsible for cisplatin resistance and the ALDH
+ BCSCs displayed higher levels of reactive oxygen species (ROS) from bulk breast cancer cells. He demonstrated that the anti-alcoholism drug disulfiram (DSF) inhibited the activity of the enzyme ALDH as well as stem cell-like properties in the ALDH
+ BCSCs. He further demonstrated that DSF treatment modulated ROS accumulation and helped to overcome cisplatin-resistance synergistically by enhancing the cytotoxic effect of cisplatin on the ALDH
+ BCSCs and their cisplatin-resistant variant sublines, indicating that DSF can sensitize CSCs to chemotherapy [
22]. Phillips et al. demonstrated that the breast cancer MCF7 cells with CD44
+CD24
− phenotype were relatively resistant to radiotherapy, generating less ROS and had decreased DNA damage in response to radiation, leading to increased cancer cell number after short courses of fractionated irradiation [
94]. Guo et al. demonstrated that DSF with Copper(Cu
2+) triggered the apoptosis of the ALDH
+ stem-like ovarian cancer cells by regulating ALDH and increasing intracellular ROS level, and enhanced the induction of apoptosis of those stem-like cells [33]. They further investigated the effect of DSF on cisplatin-resistant ovarian CSCs. It was shown that the cisplatin-resistant ovarian cancer cells remained very sensitive to DSF-induced cytotoxicity ( cisplatin-resistant ovarian CSCs vs. parental ovarian cancer cells: apoptosis and necrosis rate 60.4% vs. 20.5%); in combination with DSF and cisplatin, relatively higher apoptosis and necrosis rate was displayed in cisplatin-resistant ovarian CSCs than in their parental cells (cisplatin-resistant ovarian CSCs vs. parental ovarian cancer cells: apoptosis and necrosis rate 81.5% vs. 50.1%), identifying DSF as a potential adjuvant [32]. In additon, Yao et al. demonstrated the role of DSF as a radio-chemosensitizer in HNSCC cell lines and confirmed the therapeutic potential of DSF in patient-derived tumor transplated xennografts [28].
CSCs change their characteristics on both the genetic and protein level in respond to the various signaling networks in the TME by activating the EMT/MET process [
83]. Considering the crucial role of CSCs involved in the EMT and metastatic process, a new window may open to study the biology of metastasis with emphasis on their importance for understanding and preventing metastasis. Strategies for CSC-targeted therapies include targeting CSC markers and EMT-related markers, and modulating of intra-/extra-cellular signals that regulate EMT and CSCs, such as TGF-β, Wnt, and β-catenin [
93]. Some pre-clinical experiments targeting EpCAM
+ CSCs have been performed, i.e., Zhou et al. assessed EpCAM-targeting CAR-T cells and their killing efficiency in vitro by co-culture with colon cancer cell lines and demonstrated that EpCAM-CAR-T cells exhibited a significantly higher apoptotic effect on cancer cells compared with non-EpCAM-modified T cells [
95]. Zhang et al. showed that EpCAM-CAR-T-cell treatment significantly restrained colorectal tumor growth and formation in mouse xenograft models [
96]. In addition, the therapies targeting at CD133
+ CSCs have shown promising results either as monotherapy or by using multiple therapeutic approaches in combination with cytostatic agents [
93]. In a phase I clinical trial, the 23 patients with hepatocellular carcinoma received CD133-directed CAR-T cell infusions, and 21 patients had not developed detectable de novo lesions within the observation time frame; thereby, the trial demonstrated feasibility, safety, and efficacy of CD133-directed CAR T-cell therapy in patients [
93]. Those pre-clinical and clinical trials provide a future perspective for the cancer treatments targeting CSCs.
In conclusion, CSCs are closely involved in tumor initiation, development, metastasis, and recurrence due to their intrinsic stemness properties and capacity to undergo EMT process. Therapies fail to achieve CSCs clearance because of their reversible phenotypic plasticity. The quiescent mesenchymal CSCs stay alive during dissemination, and they can retain the capacity to re-enter proliferative epithelial state when adapting to new TME. Understanding the significance of CSCs undergoing EMT in invasion–metastasis cascade, and demonstrating how CSCs facilitate tumor metastasis remains important and necessary. By eradicating CSCs or blocking the CSC-associated EMT process to prevent metastasis, precise personalized anti-cancer treatment can be determined for the individual patients, therefore, patients will get better therapeutic efficiency with fewer side effect, less metastasis and better prognosis.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24032555