Reinventing approved therapeutic proteins for a new dose, a new formulation, a new route of administration, an improved safety profile, a new indication, or a new conjugate with a drug or a radioactive source is a creative approach to benefit from the billions spent on developing new therapeutic proteins. These new opportunities were created only recently with the arrival of Artificial Intelligence (AI)/Machine Learning (ML) tools and high throughput screening technologies. Furthermore, the complex nature of proteins offers mining opportunities that are not possible with chemical drugs; bringing in newer therapies without spending billions makes this path highly lucrative financially while serving the dire needs of humanity.
- therapeutic proteins
- recombinant proteins
- drug–antibody combinations
- immunogenicity
- artificial intelligence
1. Introduction
2. Understanding Therapeutic Proteins
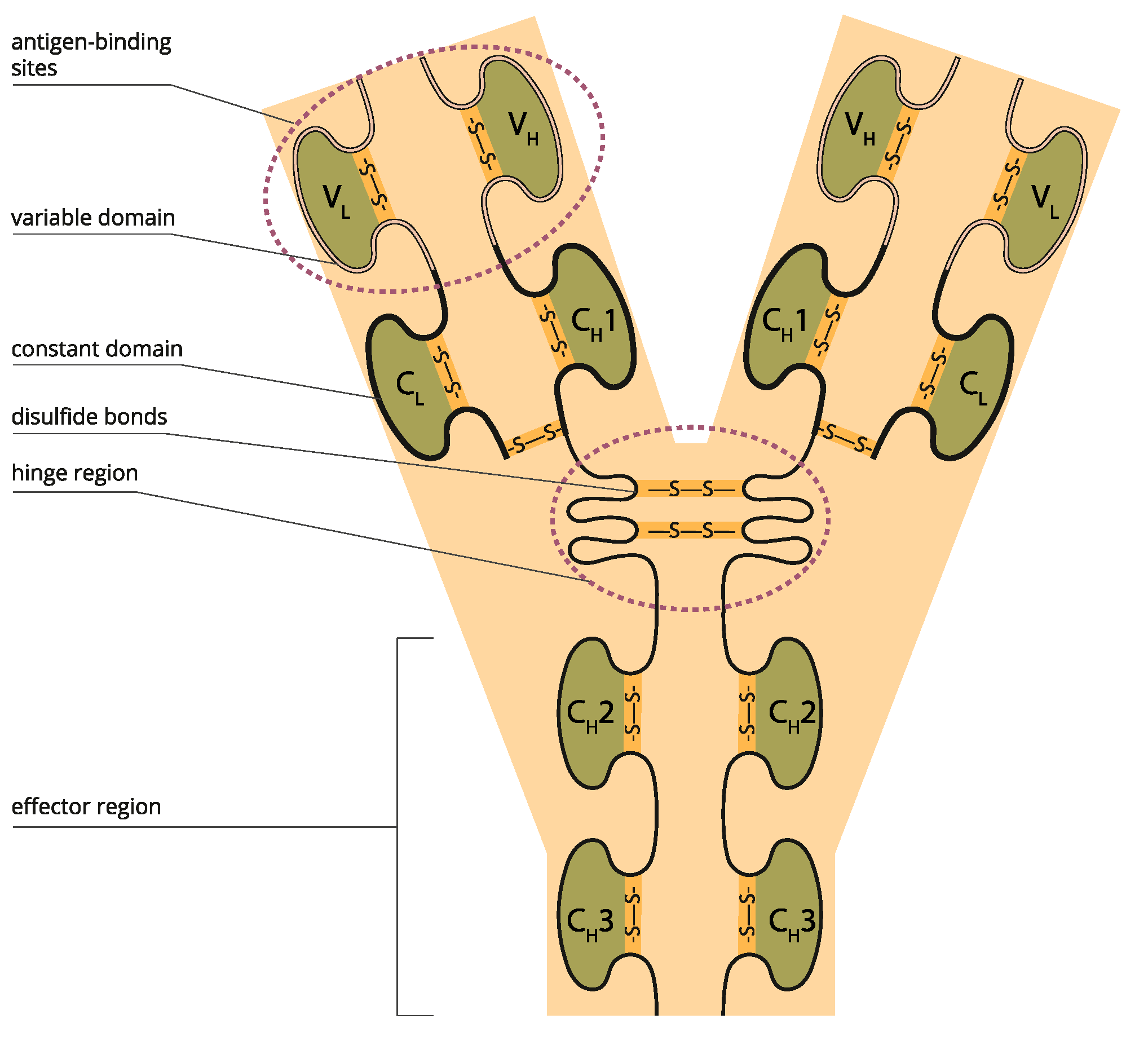
3. Reinvention Scope
4. Intellectual Property
5. Artificial Intelligence (AI) and Machine Learning (ML)
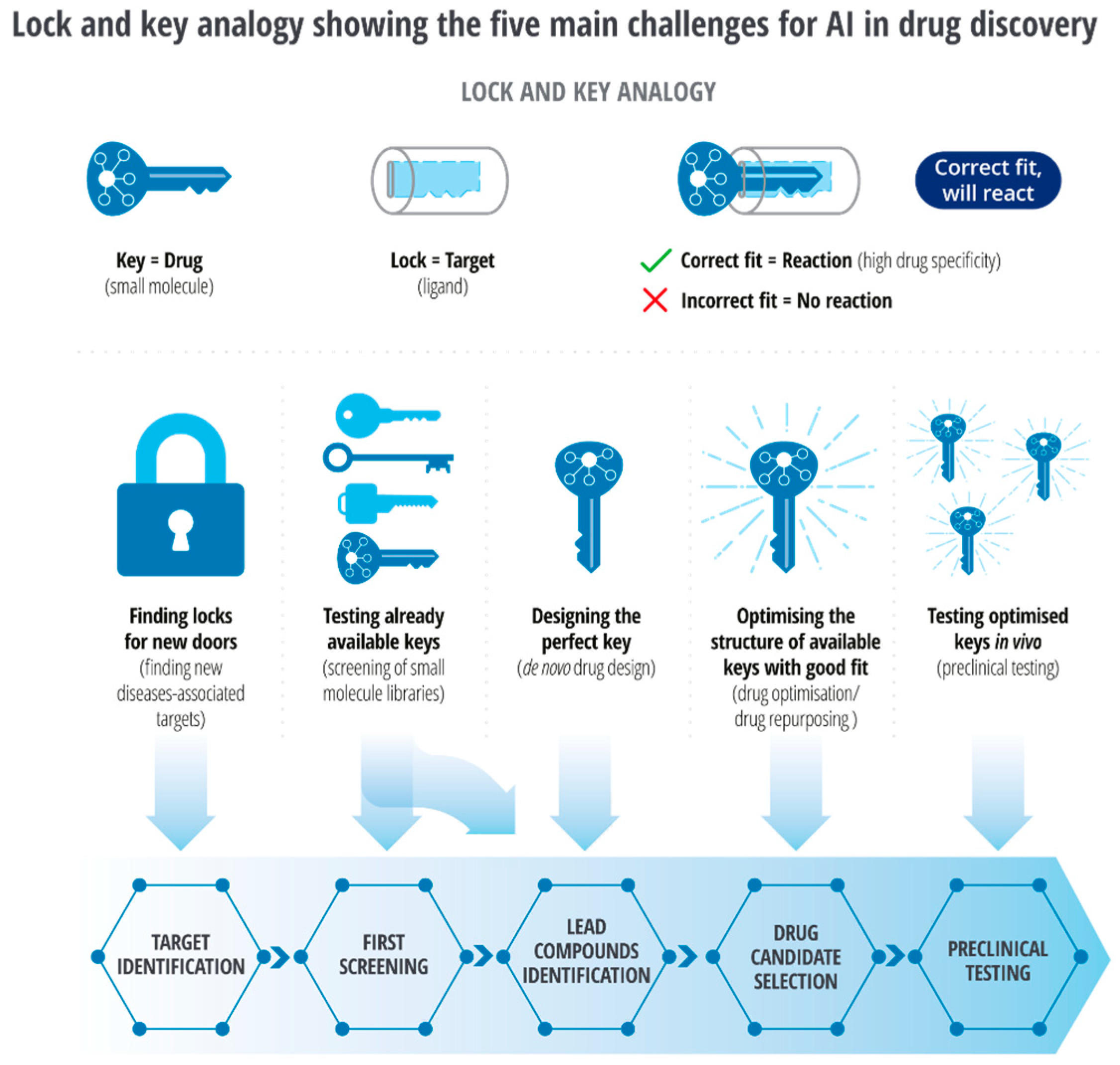
5.1. Structure Prediction
With major advancements in machine learning and AI, template-free protein structure prediction methods have also increased accuracy and reliability of structure prediction methods. Template-free AI models are trained on the sequence and structural data from openly available databases, i.e., UniProt, RCSB PDB, Uniclust [36], BFD [37], MGnify [38], etc. Highly accurate protein structure prediction tools independent of templates include AlphaFold2 [39], trTosetta [40][41], Robetta [42], RoseTTA Fold [43], ESMFold [44], and OmegaFold [45]. Each algorithm uses a different AI model to predict protein structures from amino acid sequences. For example, AlphaFold2 uses a deep neural network-based approach with over 200 million protein structures openly available in the AlphaFold2 database [46]. trRosetta uses transfer learning with pre-trained deep neural networks; the Robetta server combines ab initio and homology-based methods with machine learning algorithms.
5.2. Target Identification
5.3. Molecular Docking
5.4. Limitations
6. Structure Modifications
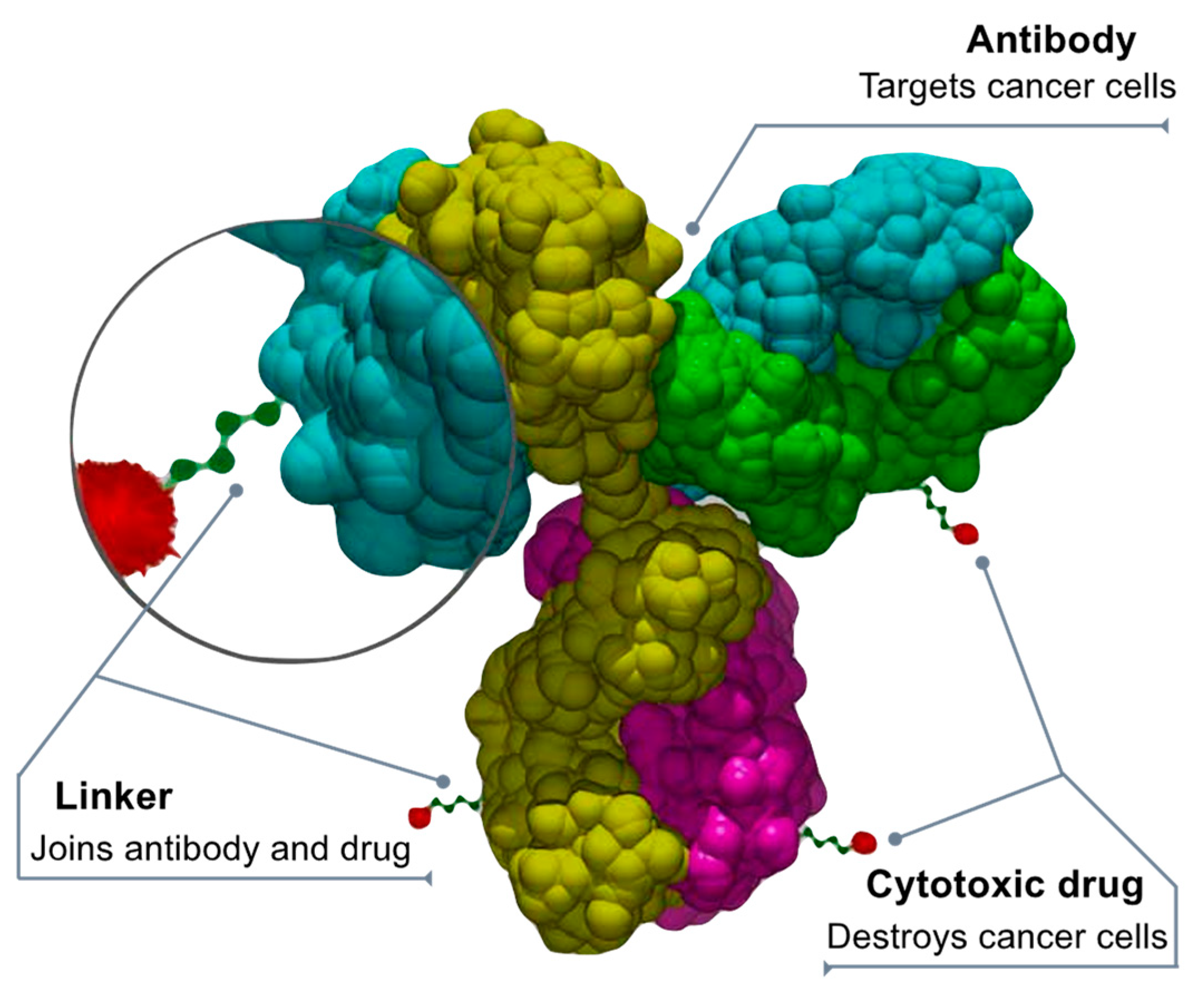
7. Drug Conjugates
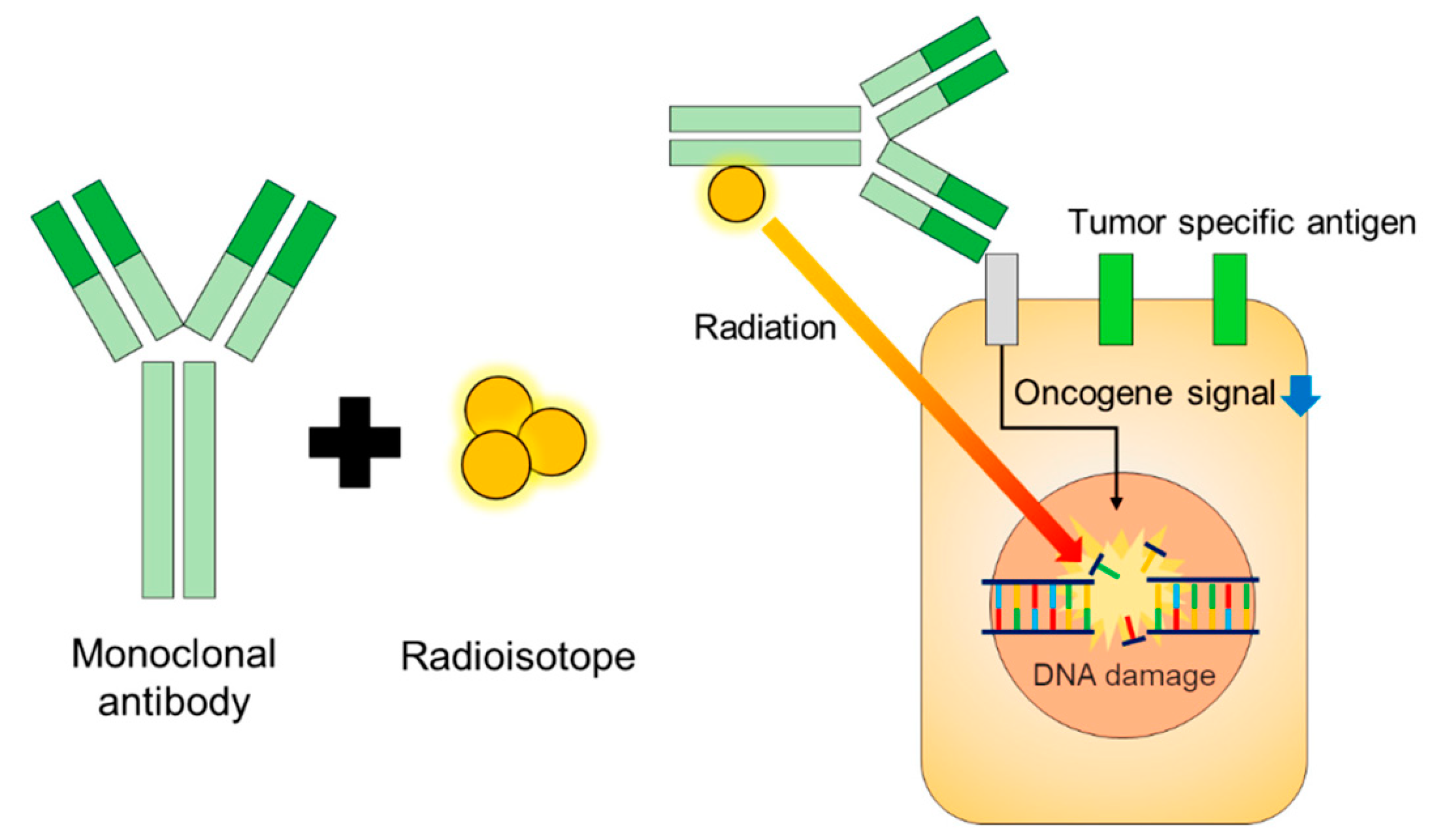
8. Radioimmunoconjugates (RIC)
9. Regulatory Perspective
10. Regulatory Submission
10.1. Nonclinical Testing
10.2. Pharmacokinetics–Pharmacodynamics
10.3. Function Testing
10.4. Immunogenic Response
This entry is adapted from the peer-reviewed paper 10.3390/biologics3020005
References
- Raju, T.N. The nobel chronicles. 1988: James Whyte Black, (b 1924), Gertrude Elion (1918-99), and George H Hitchings (1905-98). Lancet 2000, 355, 1022.
- Sean. The Process and Costs of Drug Development. FTLOScience (5 February 2023). 2022. Available online: https://ftloscience.com/process-costs-drug-development/ (accessed on 30 March 2023).
- Pearce, R.M. Chance and the prepared mind. Science 1912, 35, 941–956.
- Wermuth, C.G. Selective optimization of side activities: The SOSA approach. Drug Discov. Today 2006, 11, 160–164.
- Prosdocimi, M.; Zuccato, C.; Cosenza, L.C.; Borgatti, M.; Lampronti, I.; Finotti, A.; Gambari, R. A Rational Approach to Drug Repositioning in β-thalassemia: Induction of Fetal Hemoglobin by Established Drugs. Wellcome Open Res. 2022, 7, 150.
- Bomprezzi, R. Dimethyl fumarate in the treatment of relapsing–remitting multiple sclerosis: An overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30.
- Blair, H.A. Dimethyl fumarate: A review in moderate to severe plaque psoriasis. Drugs 2018, 78, 123–130.
- Santoro, M.G.; Carafoli, E. Remdesivir: From Ebola to COVID-19. Biochem. Biophys. Res. Commun. 2021, 538, 145–150.
- Beck, B.R.; Shin, B.; Choi, Y.; Park, S.; Kang, K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. J. 2020, 18, 784–790.
- Gilvary, C.; Elkhader, J.; Madhukar, N.; Henchcliffe, C.; Goncalves, M.D.; Elemento, O. A machine learning and network framework to discover new indications for small molecules. PLoS Comput. Biol. 2020, 16, e1008098.
- Peng, Y.; Wang, M.; Xu, Y.; Wu, Z.; Wang, J.; Zhang, C.; Liu, G.; Li, W.; Li, J.; Tang, Y. Drug repositioning by prediction of drug’s anatomical therapeutic chemical code via network-based inference approaches. Briefings Bioinform. 2021, 22, 2058–2072.
- Cong, Y.; Shintani, M.; Imanari, F.; Osada, N.; Endo, T. A New Approach to Drug Repurposing with Two-Stage Prediction, Machine Learning, and Unsupervised Clustering of Gene Expression. OMICS A J. Integr. Biol. 2022, 26, 339–347.
- Available online: https://www.drugs.com/new-indications.html (accessed on 30 March 2023).
- Jackson, D.A.; Symons, R.H.; Berg, P. Biochemical method for inserting new genetic information into DNA of Simian Virus 40: Circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proc. Natl. Acad. Sci. USA 1972, 69, 2904–2909.
- Berg, P.; Baltimore, D.; Boyer, H.W.; Cohen, S.N.; Davis, R.W.; Hogness, D.S.; Nathans, D.; Roblin, R.; Watson, J.D.; Weissman, S.; et al. Letter: Potential biohazards of recombinant DNA molecules. Science 1974, 185, 303.
- Landgraf, W.; Sandow, J. Recombinant Human Insulins—Clinical Efficacy and Safety in Diabetes Therapy. Eur. Endocrinol. 2016, 12, 12–17.
- Usmani, S.S.; Bedi, G.; Samuel, J.S.; Singh, S.; Kalra, S.; Kumar, P.; Ahuja, A.A.; Sharma, M.; Gautam, A.; Raghava, G.P.S. THPdb: Database of FDA-approved peptide and protein therapeutics. PLoS ONE 2017, 12, e0181748.
- Dimitrov, D.S. Therapeutic proteins. Methods Mol. Biol. 2012, 899, 1–26.
- Available online: https://www.biospace.com/article/biologics-market-size-to-hit-usd-719-94-billion-by-2030-/ (accessed on 30 March 2023).
- FDA. Available online: https://www.fda.gov/media/107622/download (accessed on 30 March 2023).
- Available online: https://www.ncbi.nlm.nih.gov/books/NBK562260/#:~:text=A%20peptide%20is%20a%20short,the%20building%20block%20of%20proteins (accessed on 30 March 2023).
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620.
- Lagassé, H.D.; Alexaki, A.; Simhadri, V.L.; Katagiri, N.H.; Jankowski, W.; Sauna, Z.E.; Kimchi-Sarfaty, C. Recent advances in (therapeutic protein) drug development. F1000Research 2017, 6, 113.
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug reinventing from the perspective of pharmaceutical companies. Br. J. Pharmacol. 2018, 175, 168–180.
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Kesavan, M.; Kumar, D.; Singh, R.K. Drug reinventing approach to fight COVID-19. Pharmacol. Rep. 2020, 72, 1479–1508.
- Available online: https://www.greyb.com/blog/biologics-patents-expiring-2022-2023-2024-2025-2026-2027/ (accessed on 30 March 2023).
- Goode, R.; Chao, B. Biological patent thickets and delayed access to biosimilars, an American problem. J. Law Biosci. 2022, 9, lsac022.
- Beneke, F.; Mackenrodt, M.-O. Artificial intelligence and collusion. IIC Int. Rev. Intellect. Prop. Compet. Law 2019, 50, 109–134.
- Bielecki, A.; Bielecki, A. Foundations of artificial neural networks. In Models of Neurons and Perceptrons: Selected Problems and Challenges; Janusz, K., Ed.; Springer International Publishing; Polish Academy of Sciences: Warsaw, Poland, 2019; pp. 15–28.
- Da Silva, I.N. Artificial Neural Networks; Springer: Berlin/Heidelberg, Germany, 2017.
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of artificial intelligence for computer-assisted drug discovery. Chem. Rev. 2019, 119, 10520–10594.
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity prediction using deep learning. Front. Environ. Sci. 2016, 3, 80.
- Moll, S.; Desmoulière, A.; Moeller, M.J.; Pache, J.-C.; Badi, L.; Arcadu, F.; Richter, H.; Satz, A.; Uhles, S.; Cavalli, A.; et al. DDR1 role in fibrosis and its pharmacological targeting. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 118474.
- Ren, F.; Ding, X.; Zheng, M.; Korzinkin, M.; Cai, X.; Zhu, W.; Mantsyzov, A.; Aliper, A.; Aladinskiy, V.; Cao, Z.; et al. AlphaFold accelerates artificial intelligence powered drug discovery: Efficient discovery of a novel CDK20 small molecule inhibitor. Chem. Sci. 2023, 14, 1443–1452.
- Deloitte—Intelligent Drug Discovery. (n.d.). Deloitte. Available online: https://www2.deloitte.com/content/dam/Deloitte/my/Documents/risk/my-risk-sdg3-intelligent-drug-discovery.pdf (accessed on 8 March 2023).
- Mirdita, M.; Driesch, L.V.D.; Galiez, C.; Martin, M.-J.; Söding, J.; Steinegger, M. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 2017, 45, D170–D176.
- BFD. (n.d.). Available online: https://bfd.mmseqs.com/ (accessed on 30 March 2023).
- Mitchell, A.L.; Scheremetjew, M.; Denise, H.; Potter, S.; Tarkowska, A.; Qureshi, M.; A Salazar, G.; Pesseat, S.; A Boland, M.; Hunter, F.; et al. EBI Metagenomics in 2017: Enriching the analysis of microbial communities, from sequence reads to assemblies. Nucleic Acids Res. 2018, 46, D726–D735.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589.
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503.
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651.
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531.
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876.
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv 2022.
- Wu, R.; Ding, F.; Wang, R.; Shen, R.; Zhang, X.; Luo, S.; Su, C.; Wu, Z.; Xie, Q.; Berger, B.; et al. High-resolution de novo structure prediction from primary sequence. bioRxiv 2022.
- Database, A.P.S. (n.d.). AlphaFold Protein Structure Database. Available online: https://alphafold.ebi.ac.uk/ (accessed on 23 March 2023).
- Yang, C.; Chen, E.A.; Zhang, Y. Protein–Ligand Docking in the Machine-Learning Era. Molecules 2022, 27, 4568.
- de Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11.
- Lamberti, M.J. A study on the application and use of artificial intelligence to support drug development. Clin. Ther. 2019, 41, 1414–1426.
- Choi, S.; Park, H.; Jung, S.; Kim, E.-K.; Cho, M.-L.; Min, J.-K.; Moon, S.-J.; Lee, S.-M.; Cho, J.-H.; Lee, D.-H.; et al. Therapeutic Effect of Exogenous Truncated IK Protein in Inflammatory Arthritis. Int. J. Mol. Sci. 2017, 18, 1976.
- Rigi, G.; Kardar, G.; Hajizade, A.; Zamani, J.; Ahmadian, G. The effects of a truncated form of Staphylococcus aureus protein A (SpA) on the expression of cytokines of autoimmune patients and healthy individuals. Europe PMC, 2022; not peer-reviewed.
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. Available online: https://pubmed.ncbi.nlm.nih.gov/26182432/ (accessed on 23 March 2023).
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19.
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 1–25.
- Jaffray, D.A. Image-guided radiotherapy: From current concept to future perspectives. Nat. Rev. Clin. Oncol. 2012, 9, 688–699.
- Nasr, D.; Kumar, P.A.; Zerdan, M.B.; Ghelani, G.; Dutta, D.; Graziano, S.; Lim, S.H. Radioimmunoconjugates in the age of modern immuno-oncology. Life Sci. 2022, 310, 121126.
- Pouget, J.P.; Constanzo, J. Revisiting the radiobiology of targeted alpha therapy. Front. Med. 2018, 8, 692436.
- Grillo-López, A.J. Zevalin: The first radioimmunotherapy approved for the treatment of lymphoma. Expert. Rev. Anticancer Ther. 2002, 2, 485–493.
- Zaheer, J.; Kim, H.; Lee, Y.-J.; Kim, J.S.; Lim, S.M. Combination Radioimmunotherapy Strategies for Solid Tumors. Int. J. Mol. Sci. 2019, 20, 5579.
- Miranda, A.C.C.; Santos, S.N.D.; Fuscaldi, L.L.; Balieiro, L.M.; Bellini, M.H.; Guimarães, M.I.C.C.; de Araújo, E.B. Radioimmunotheranostic pair based on the anti-HER2 monoclonal antibody: Influence of chelating agents and radionuclides on biological properties. Pharmaceutics 2021, 13, 971.
- U S Food and Drug Administration. Clinical Pharmacology Review for Application 214787Orig1S000 (Remdesivir). 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/214787Orig1s000ClinpharmR.pdf (accessed on 23 March 2023).
- Lim, S.H.; Kim, K.; Choi, C.-I. Pharmacogenomics of Monoclonal Antibodies for the Treatment of Rheumatoid Arthritis. J. Pers. Med. 2022, 12, 1265.
- Niazi, S. Volume of Distribution as a Function of Time. J. Pharm. Sci. 1976, 65, 452–454.
- Wesolowski, C.; Wesolowski, M.J.; Babyn, P.S.; Wanasundara, S.N. Time Varying Apparent Volume of Distribution and Drug Half-Lives Following Intravenous Bolus Injections. PLoS ONE 2016, 11, e0158798.
- Gadkar, K.; Yadav, D.B.; Zuchero, J.Y.; Couch, J.A.; Kanodia, J.; Kenrick, M.K.; Atwal, J.K.; Dennis, M.S.; Prabhu, S.; Watts, R.J.; et al. Mathematical PKPD and safety model of bispecific TfR/BACE1 antibodies for the optimization of antibody uptake in brain. Eur. J. Pharm. Biopharm. 2016, 101, 53–61.
- Wittrup, K.D.; Thurber, G.M.; Schmidt, M.M.; Rhoden, J.J. Practical theoretic guidance for the design of tumor-targeting agents. Methods Enzymol. 2012, 503, 255–268.
- Yeung, Y.A.; Leabman, M.K.; Marvin, J.S.; Qiu, J.; Adams, C.W.; Lien, S.; Starovasnik, M.A.; Lowman, H.B. Engineering human IgG1 affinity to human neonatal Fc receptor: Impact of affinity improvement on pharmacokinetics in primates. J. Immunol. 2009, 182, 7663–7671.
- Deng, R.; Loyet, K.M.; Lien, S.; Iyer, S.; Deforge, L.E.; Theil, F.-P.; Lowman, H.B.; Fielder, P.J.; Prabhu, S. Pharmacokinetics of humanized monoclonal anti-tumor necrosis factor-α antibody and its neonatal Fc receptor variants in mice and cynomolgus monkeys. Drug Metab. Dispos. 2010, 38, 600–605.
- McClellan, J.E.; Conlon, H.D.; Bolt, M.W.; Kalfayan, V.; Palaparthy, R.; Rehman, M.I.; Kirchhoff, C.F. The ‘totality-of-the-evidence’ approach in the development of PF-06438179/GP1111, an infliximab biosimilar, and in support of its use in all indications of the reference product. Ther. Adv. Gastroenterol. 2019, 12, 1756284819852535.
- Ryding, J.; Stahl, M.; Ullmann, M. Demonstrating biosimilar and originator antidrug antibody binding comparability in antidrug antibody assays: A practical approach. Bioanalysis 2017, 9, 1395–1406.
- Wang, X.; An, Z.; Luo, W.; Xia, N.; Zhao, Q. Molecular and functional analysis of monoclonal antibodies in support of biologics development. Protein Cell 2018, 9, 74–85.
- Todoroki, K.; Yamada, T.; Mizuno, H.; Toyo’Oka, T. Current Mass Spectrometric Tools for the Bioanalyses of Therapeutic Monoclonal Antibodies and Antibody-Drug Conjugates. Anal. Sci. 2018, 34, 397–406.
- Láng, J.A.; Balogh, Z.C.; Nyitrai, M.F.; Juhász, C.; Gilicze, A.K.B.; Iliás, A.; Zólyomi, Z.; Bodor, C.; Rábai, E. In vitro functional characterization of biosimilar therapeutic antibodies. Drug Discov. Today Technol. 2020, 37, 41–50.
- Cymera, F.; Becka, H.; Rohde, A.; Reusch, D. Therapeutic monoclonal antibody N-glycosylation—Structure, function and therapeutic potential. Biologicals 2018, 52, 1–11.
- Prior, S.; Hufton, S.E.; Fox, B.; Dougall, T.; Rigsby, P.; Bristow, A. Participants of the study International standards for monoclonal antibodies to support pre- and post-marketing product consistency: Evaluation of a candidate international standard for the bioactivities of rituximab. Mabs 2018, 10, 129–142.