Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Doaa Kirat and Version 2 by Sirius Huang.
Physiologically, autophagy is an evolutionarily conserved and self-degradative process in cells. Autophagy carries out normal physiological roles throughout mammalian life. Accumulating evidence shows autophagy as a mechanism for cellular growth, development, differentiation, survival, and homeostasis.
- bulk autophagy
- selective autophagy
- testis
- reproductive physiology
1. Introduction
In 1963, the word autophagy was coined by the Belgian cytologist and biochemist Christian de Duve who was rewarded the Nobel Prize in Physiology or Medicine in 1974 for his discovery of lysosomes and peroxisomes. In the 1990s, Japanese biologist Yoshinori Ohsumi identified autophagy-related genes. In 2016, Ohsumi won the Nobel Prize in Physiology or Medicine for his discovery of the molecular mechanisms of autophagy. Ohsumi’s discoveries opened the way to recognize the fundamental significance of autophagy in many physiological processes.
Autophagy, a lysosome-mediated intracellular degradation pathway, is an evolutionarily conserved mechanism in eukaryotes. Autophagy is a Greek word where “auto” means self and “phagy” means eating. This implies that autophagy is a process in which the cell eats its own components, similarly to cellular cannibalism.
Autophagy plays fundamental roles in numerous physiological processes [1]. The physiological roles of autophagy are eliminating unnecessary cargoes, sequestering organelles, recycling cellular components, controlling organelle homeostasis, promoting cell survival, and providing required resources [2]. Autophagy is an intracellular degradation process in which unwanted cargoes, such as old or damaged organelles, and unneeded proteins are sequestrated into double-membrane vesicles called autophagosomes and subsequently delivered to the lysosomes for degradation by lysosomal hydrolases [3]. The macromolecular contents from this digestion are released back into the cytosol in order to be reused for cellular and tissue remodeling [3]. Therefore, the catabolic role of the autophagy pathway allows various cell types to maintain and control cellular homeostasis, renew the cells, and provide energy [4]. Consequently, the dysregulation and dysfunction of autophagy are implicated in various types of diseases, such as neurodegenerative diseases (Alzheimer’s, Huntington’s, and Parkinson’s disease) and tumorigenesis [5][6][5,6].
In mammalian species, there are three types of autophagy—macroautophagy, microautophagy, and chaperone-mediated autophagy—and all of them promote the proteolytic degradation of cytosolic components and cargoes in the lysosome and the reuse of synthesized macromolecules by the cell [7]. Different types of autophagy share the common feature of the lysosomal degradation of damaged proteins but differ in their mechanisms of delivering the substrate to the lysosome [8]. Macroautophagy depends on the formation of autophagosomes in order to transport cargo to the lysosome [9], while micro-autophagy involves the direct uptake of cargo via the invagination of the lysosomal membrane [10]. Meanwhile, chaperone-mediated autophagy-targeted proteins are translocated across the lysosomal membrane in a complex with chaperone proteins that are recognized by specific-receptor-lysosomal-associated membrane proteins [10][11][10,11]. Neither microautophagy nor chaperone-mediated autophagy involves autophagosome formation but instead depends on the degradation function of lysosomes.
2. Mammalian Autophagy Machinery and Autophagy-Related Genes
Briefly, the process of macroautophagy includes five stages: initiation, elongation, maturation, fusion, and degradation. In mammals, the process of autophagy initiates the formation of intracellular membrane-bounded organelles enriched in phosphatidylinositol 3-phosphate, known as omegasomes, that is dynamically connected to the endoplasmic reticulum [12][23]. The small cup-shaped membrane structure termed phagophore is de novo formed from omegasomes. After that, this phagophore undergoes nucleation followed by elongation to engulf cargoes and close to form autophagosomes [13][14][24,25]. Autophagy is terminated with the fusion of the autophagosome with the lysosome to form an autolysosome with the subsequent degradation of autolysosomal content by lysosomal hydrolases [15][26]. The resulting simple molecules, including free fatty acids, amino acids, and nucleotides, are recycled back to the cytosol by lysosomal permease and reused as an energy source by the cell [16][27]. Autophagy is a complex process that is regulated by a series of protein complexes and signaling pathways. In mammals, the core autophagy-related (ATG) genes and their protein products are generally classified into the ULK1 protein kinase complex [17][28], Vps34-beclin1 class III PI3-kinase complex [18][19][29,30], ATG9A transportation system [20][21][31,32], ATG12 conjugation system [22][33], and LC3 conjugation system [23][34]. Table 12 illustrates the components and role of autophagy complexes participating in the mammalian autophagy machinery.Table 12.
Components and role of autophagy complexes participating in the mammalian autophagy machinery.
| Complex | Core Components | Autophagic Role |
|---|---|---|
| ULK1/2 complex | ULK1/2 ATG13 FIP200 ATG101 |
Initiation |
| BECN1 complex | Beclin1 VPS34 VPS15 ATG14L |
Nucleation |
| ATG9A complex | ATG9A WIPI1/2 ATG2A |
Initiation |
| Ubiquitin-like complex | LC3A-C, GABARAP | Cargo selection and Elongation |
| ATG12 ATG4 ATG7 ATG3 ATG10 ATG5 ATG16L1 |
Elongation |
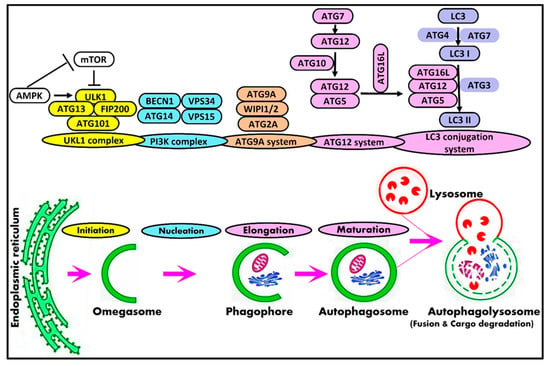
Figure 1.
Schematic representation of macroautophagy pathway and core autophagy-related proteins in mammals.
Potential Mechanisms of Selective Macroautophagy
The selectivity of autophagy is a prevalent phenomenon in various cells. Current research has proven the presence of various kinds of selective autophagy in eukaryotic cells. According to different cargoes, selective autophagy can be divided into several subcategories, such as mitophagy, proteaphagy, ribophagy, pexophagy, lysophagy, and nucleophagy [65][76]. Selective autophagy is principally dependent on both the recognition of the cargo and the coupling of the cargo to the phagophore, which can be carried out by proteins called selective autophagy receptors or cargo receptors [66][77]. A representative overview of the mechanisms of selective macroautophagy is illustrated in Figure 2. The first mechanism involves selective autophagy receptors that act as a bridge between the phagophore and cargo to facilitate the recruitment of autophagic machinery, mainly by the binding of LC3 and then the degradation of the cargo [67][78].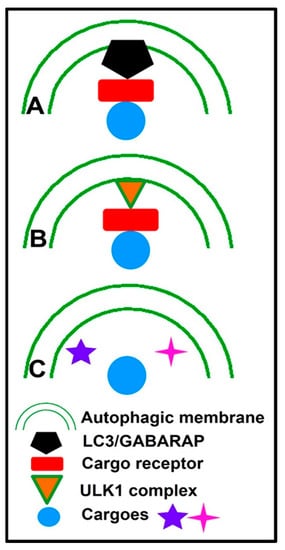
Figure 2.
Representative overview of the mechanisms of selective macroautophagy.