1. Sirt3 and Alzheimer’s Disease
Alzheimer’s disease (AD), also known as senile dementia, is a neuronal degenerative disease that occurs mainly in the elderly and is characterized clinically by cognitive dysfunction, progressive memory impairment, language impairment, and personality changes
[1][103]. Studies have shown that the pathogenesis of AD involves mitochondrial dysfunction, which is caused by multiple determinants that ultimately lead to necrosis, neuronal degeneration, or apoptosis
[2][1]. Sirt3, an important regulator of protein deacetylation in the cellular mitochondria, plays a key role in maintaining the functional integrity of the mitochondria and has therefore received increasing attention in the study of AD. The current mechanisms of Sirt3 in AD mainly include (1) increasing the ATP levels in the mitochondria and promoting mitochondrial biosynthesis
[3][104], (2) activating and enhancing mitochondrial dynamics
[4][105], and (3) counteracting oxidative stress and regulating neuronal excitability
[5][106]. Consequently, Sirt3 plays a protective role in AD.
Notably, the neurotoxic effects of amyloid beta (Aβ) play a crucial role in the development of AD
[6][107]. In cortical samples from AD patients, the expression of Sirt3 mRNA is lower than that in healthy people
[7][76]. According to a past study, neuronal apoptosis occurred when Aβ was added to a primary neuronal culture model, which was reversed using the neuroprotective factor pituitary adenosine live cell peptide (PACAP), whose protective effect was associated with the activation of mitochondrial Sirt3 synthesis
[8][108]. After the knockdown of Sirt3, PACAP-mediated neuroprotection was lost
[9][109]. In addition, curcumin, which has neuroprotective effects, attenuated Aβ-induced neuronal metabolic dysfunction and improved cognitive performance in a mouse model of AD by increasing Sirt3 activity
[10][79]. This suggests that Sirt3 may have a neuroprotective effect on AD by modulating Aβ.
In addition to Aβ deposition, apolipoprotein E4 (APOE4) is an important genetic factor associated with the late onset of AD. Impaired learning and memory were observed in APOE4 transgenic mice
[11][12][110,111]. In a study on human beings, Sirt3 expression was downregulated in the cerebral cortex of the APOE4 group compared to those without APOE4 expression
[7][76]. APOE4 expression triggers mitochondrial oxidative stress, reduces ATP synthesis, leads to mitochondrial dysfunction, and subsequently disrupts synaptic transmission and leads to the emergence of cognitive impairment
[13][77]. Meanwhile, the overexpression of Sirt3 improves memory and learning in APOE4 transgenic mice
[14][78], which may be related to the fact that Sirt3 improves the antioxidant capacity of the nerve cells
[15][112]. By regulating APOE4 and by improving brain energy metabolism, Sirt3 again plays a role with neuroprotective effects.
Thus, Sirt3 is crucial in AD-related pathogenesis (Table 1). Overall, as the world’s aging population continues to evolve, the Sirt3-mediated protective mechanisms provide an adequate basis for Sirt3 as a therapeutic target for AD, which can effectively prevent AD, can reduce the social burden, can improve the quality of life, and will become a potential target for the treatment of neurodegenerative diseases.
Table 1.
Sirt3′s roles in neurodegenerative diseases.
| Neurodegenerative Disease |
Mechanism |
Experimental Setting |
Research |
| AD |
ROS in mitochondria increase Sirt3 expression. |
Cell model |
[16][73] |
| Pharmacological enhancement of mitochondrial ROS increases the expression of Sirt3 in primary hippocampal cultures. |
AD mouse model and cell model |
[17][74] |
| PACAP stimulates the production of mitochondrial Sirt3 and reduces neuronal death. |
Postmortem human tissue, triple transgenic mouse model, and cell model |
[18][75] |
| Amyloid-β increases levels of total tau and acetylated tau through its modulation of Sirt3. |
Postmortem human tissue |
[7][76] |
| APOE4 reduces ATP production by modulating the PGC-1α-Sirt3 signaling pathway, triggering mitochondrial oxidative stress and disrupting synaptic function. |
Postmortem human tissue |
[13][77] |
| Sirt3 may mediate the neuroprotection of ketones by increasing neuronal energy metabolism. |
APOE4 mouse model |
[14][78] |
| Alleviation of Aβ 42-induced neuronal metabolic dysfunction occurs via the THRB/Sirt3 axis and improves cognition. |
APPTG mouse model |
[10][79] |
| Activation of mitophagy and mitochondrial unfolded protein response occurs. |
APP/PS1 mouse model |
[19][80] |
| PD |
IC87201 and ZL006 reduce ROS production and improve mitochondrial dysfunction by increasing the expression of Sirt3 after MPP+ exposure. |
MPP+-induced primary cortical neuron cell models |
[20][81] |
| Sirt3 has a possible role in MPTP-induced neurodegeneration by preserving the free radical scavenging capacity of mitochondria. |
Sirt3 null mouse model |
[21][82] |
| Sirt3 overexpression dramatically increases cell viability, decreases cell apoptosis, prevents the accumulation of α-synuclein, suppresses the reduction of SOD and glutathione, decreases ROS generation, and alleviates MMP collapse induced by rotenone. |
PD cell model |
[22][83] |
| Sirt3 rescues neurons through the stabilization of mitochondrial biogenetics. |
Virally expressed mutant α-synuclein rat model of parkinsonism |
[23][84] |
| Curcumin lowers ROS levels in SH-SY5Y cells and upregulates Sirt3 expression. |
SH-SY5Y cell models |
[24][85] |
| miR-494-3p downregulation increases Sirt3 expression, reduces oxidative stress, and improves dyskinesia. |
MPTP-induced PD mouse model and SH-SY5Y cell model |
[25][86] |
| Saikosaponin-d exerts a neuroprotective effect by upregulating Sirt3 expression and alleviating oxidative stress damage. |
MPP+-induced SH-SY5Y cell models |
[26][87] |
| Sirt3 mediates SOD2 deacetylation to reduce ROS accumulation and to restore mitochondrial function, thereby preventing apoptosis. |
6-OHDA-treated rat, MPTP-treated mouse, and zebrafish models |
[27][88] |
| Regulation of Sirt3 in mitochondrial functions and oxidative stress occurs in PD. |
Sirt3 null mouse and PD mouse models |
[28][89] |
| Upregulated Sirt3 mitigates the protective effect of mitochondrial dysfunction on neuronal damage. |
SH-SY5Y cell models |
[29][90] |
| HD |
Knockdown of Sirt3 significantly inhibits viniferin-mediated AMP-activated kinase activation and diminishes the neuroprotective effects of viniferin. |
Mutant HTT cell model |
[30][91] |
| Increased Sirt3 levels and/or activity reduce oxidative damage. |
Cell model, HD knockin mouse model, and Huntington’s disease transgenic (YAC128) mouse model |
[31][32][33][92,93,94] |
| Sirt3 protects neurons against metabolic and oxidative stress by reducing mitochondrial superoxide levels, stabilizing cellular and mitochondrial Ca2+ homeostasis, and inhibiting mitochondrial membrane permeability transition pore formation to prevent apoptosis. |
Cell model and HD mouse model |
[34][95] |
| Sirt3 overexpression promotes the antioxidant effect of cells expressing mutant HTT, leading to enhanced mitochondrial function and balanced dynamics. |
Postmortem human tissue and primary striatal neuron cell model |
[35][96] |
| ALS |
Sirt3 protects against mitochondrial fragmentation and neuronal cell death with mutant SOD1 (G93A). |
SOD1G93A transgenic mouse model and primary cortical neuronal cell model |
[36][97] |
| Overexpression of Sirt3 increases NADPH levels and protects from oxidative-stress-induced cell death. |
Sirt3 mouse model |
[37][98] |
| Grape wine polyphenols prevent axonal apoptosis and act via mitochondrial Sirt3 activation in axons. |
Primary cortical neuronal cell model |
[38][99] |
| Sirt3 can restore neuronal mitochondrial fragmentation and transport disorders, reducing neuronal death, and protects against mitochondrial alterations. |
SOD1-mutant cell model |
[39][100[40],101] |
| MS |
The EA protects muscle tissue from cuprizone-induced demyelination by overexpressing Sirt3 to protect mitochondria and to reduce oxidative stress. |
Mouse model |
[41][102] |
2. Sirt3 and Parkinson’s Disease
As the second-most prevalent neurodegenerative condition in the world, PD typically affects people over the age of 65, significantly impairs their ability to move, and impacts the quality of their lives
[42][113]. Numerous investigations have suggested that dopaminergic neuron denaturation and death in the substantia nigra can be caused by mitochondrial malfunction
[43][114], oxidative stress
[44][115], anomalies in the ubiquitin–protease system, and α-synuclein accumulation
[45][116]. A large number of studies have started to place a great emphasis on the connection between Sirt3 and PD’s pathogenesis because of the significant roles that energy metabolism disorders, mitochondrial oxidative stress, and the PD susceptibility genes PARKIN and PINK1 play in maintaining mitochondrial homeostasis
[28][89].
The pathogenesis of PD is significantly influenced by mitochondrial dysfunction. In rat models of PD, abnormally folded and aggregated α-synuclein activates oxidative stress, damaging the mitochondria, which, in turn, harms neurons
[45][116]. Recently, it was reported that Sirt3 protects the neurons by stabilizing mitochondrial energy metabolism in PD
[23][84]. Sirt3 attenuates the death of the nigra dopaminergic neurons by reducing the buildup of oxidative stress products by deacetylating SOD2 and ATP synthase β-subunit
[21][82]. Another study found that theacrine, a purine alkaloid, inhibits ROS production by activating mitochondrial Sirt3 and that it ultimately inhibits the apoptosis of the dopaminergic neurons
[27][88]. As claimed by some researchers, a Sirt3 knockdown significantly exacerbates the death of the neurons and increases α-synuclein accumulation, whereas Sirt3 overexpression substantially reduces apoptosis, enhances cell viability, blocks the accumulation of α-synuclein, and decreases ROS production
[22][25][83,86].
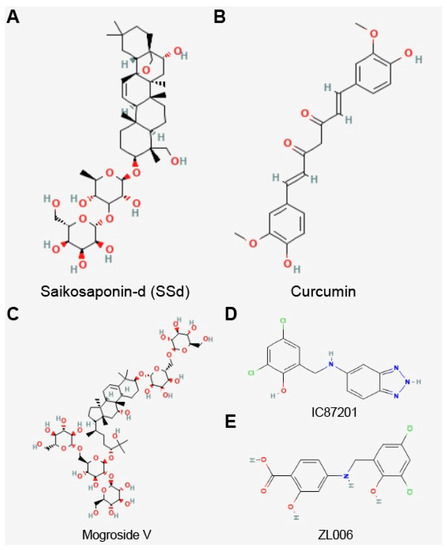
Currently, a connection between Sirt3 and PD has been discovered in a few drug studies. The neuroprotective effect of saikosaponin-d (SSd)
[26][87], curcumin
[24][85], and mogroside V
[29][90] in PD cell models may be related to the reduction in the ROS level and the upregulation of Sirt3 expression (
Figure 14). The compounds IC87201 and ZL006 activate the expression of Sirt3 through the inhibition of the interaction between postsynaptic density protein 95 (PSD-95) and neuronal nitric oxide synthases (nNOS), thereby mitigating the neuronal toxicity of PD
[20][81]. In addition, melatonin mitigates PD dopaminergic neuronal damage by upregulating Sirt3 expression. Its mechanism of action is linked to its inhibition of microglia activation, alleviating inflammatory damage and oxidative stress.
Figure 14.
Chemical structure of Sirt3 activators. (
A
) Saikosaponin-d (SSd), (
B
) curcumin, (
C
) mogroside V, (
D
) IC87201, and (
E
) ZL006.
The above findings reveal that Sirt3 has a certain relationship with the occurrence of PD, and Sirt3 can become a new target for PD therapeutic interventions; however, the investigation of the molecular mechanism of Sirt3’s specific role in PD development is not profound enough. Therefore, further studying the protective mechanism of Sirt3 in PD is crucial.
3. Sirt3 and Huntington’s Disease
HD is a rare autosomal dominant hereditary neurodegenerative illness characterized by progressive aggravated extrapyramidal symptoms, cognitive impairment, behavioral problems, and persistent chorea-like movements. The loss of many spiny striatal efferent neurons in the basal ganglia region, which results in aberrant dopamine, glutamic acid, and γ-aminobutyric acid transmission, is the primary pathogenic manifestation of HD
[46][117]. As abnormal Huntingtin (Htt), a protein associated with HD, formation occurs with polyglutamine, patients develop metabolic disorders which may be caused by mitochondrial dysfunction
[47][118]. Sirt3 has been reported to control mitochondrial function and is linked to oxidative damage, and it may provide a new biological target for HD treatment
[48][119].
Studies on animals and people have discovered that the use of mitochondrial-oriented antioxidants in the treatment of oxidative damage can affect the level of Sirt3
[35][96], indicating that changes in the level and/or activity of Sirt3 are responses to significant oxidative damage
[49][50][8,120]. A clear example is that the expression level of Sirt3 in the neurons with abnormal Htt in HD models is significantly reduced
[30][91]. Additionally, trans-ε-viniferin can maintain the expression of Sirt3 in the cells, mediate the activation of AMPK and SOD2, alleviate the accumulation of ROS in the cells, promote the biogenesis of the mitochondria, improve the survival rate of the HD striatal cells, and produce neuroprotective effects
[30][51][91,121]. Moreover, when using the HD mouse model induced with 3-nitropropionic acid (3-NP), it was found that Sirt3 knockout mice are more susceptible to the toxic effect of 3-NP than wild-type mice, further pointing out that Sirt3 might be an essential target in HD therapy
[34][95].
This suggests that Sirt3 has a certain relationship with the occurrence of HD; however, clarifying the function of Sirt3 still necessitates more in-depth research and exploration. The study of Sirt3 will offer a novel approach for HD treatment due to the imbalance of Sirt3 expression.
4. Sirt3 and Amyotrophic Lateral Sclerosis
The majority of ALS patients die of respiratory failure or paralysis three to five years after the onset of symptoms
[52][122]. ALS is a progressive lesion that causes motor neuron destruction in the anterior horn of the spinal cord
[53][123]. The currently recognized pathogenesis of ALS consists of prion-like proliferation, an imbalance of protein homeostasis in the CNS, mitochondrial dysfunction, the spread of abnormal proteins, glutamate-mediated excitatory neurotoxicity, intraneuronal substance transport disorders, RNA metabolic disorders, and the abnormal apoptosis of the neurons
[54][124]. The metabolic master regulator PGC-1α, a moderator of ALS in humans and model species, regulates Sirt3 expression
[55][125]. Sirt3 reverses the abnormal metabolic patterns in the ALS motor neurons by acting as a mitochondrial deacetylase in ALS patients, preserving mitochondrial function and integrity
[56][126].
Recent research has shown that boosting NAD
+ levels, Sirt3 activity, and antioxidant defenses may be effective treatments for ALS
[57][58][127,128]. The first evidence of a protective function of the Sirt3 single-nucleotide polymorphism rs4980329 in ALS came from a genetic investigation
[59][129]. Mutations in the SOD1 gene can result in the hereditary neurodegenerative disease ALS
[60][130] since it causes the shortening of the mitochondria, as well as an increase in rounded, fragmented mitochondria, affecting transport and ultimately leading to motor neuron death in the spinal cord
[61][131]. Sirt3 can restore neuronal mitochondrial fragmentation and transport disorders caused by SOD1 mutations to a certain extent, reducing neuronal death and protecting against mitochondrial alterations in the SOD1-mutant neurons
[36][97]. Moreover, Sirt3 can effectively antagonize SOD1-mutant astrocyte-mediated motor neuron damage, providing a new direction for the treatment of ALS
[40][101].
At present, there is no effective cure for ALS, and the treatment is currently based on delaying the disease’s progress and improving the patient’s quality of life. With the deepening of the study of the Sirt3 mechanism, plenty of investigations have proven that Sirt3 is closely associated with ALS. Even though the specific relationship cannot be determined, it also provides a new direction for Sirt3-targeted formulations for ALS patients’ treatment.
5. Sirt3 and Multiple Sclerosis
MS is an autoimmune-mediated chronic inflammatory disease based on demyelinating CNS lesions with the pathological features of alternating relapses and remissions and the progressive loss of the neuronal myelin sheaths
[62][63][132,133]. MS mainly develops in young adults, and elderly patients tend to have a progressive disease
[64][134]. Previous research has shown that postmortem MS brain lesions and the experimental autoimmune encephalomyelitis (EAE) model both exhibit reduced Sirt2 expression
[65][135]. Similar results were found for Sirt3, which exhibits lower expression in the postmortem brain tissues of MS patients
[66][136].
A recent study showed that honokiol, a Sirt3 activator, protects C57BL/6 mice against EAE and that this protection is linked to a decrease in demyelination
[67][137]. In addition, it is thought that the Sirt3 enzyme contributes to the development of myelin sheaths around mouse brain neurons. According to these preliminary findings, Sirt3 enzymes may become prospective therapeutic targets for the treatment of mitochondrial diseases, such as MS
[68][138].
MS is associated with systemic immune disorders, the etiology of which is unclear. There are currently few studies on MS and Sirt3 (
Table 1). Future studies on MS can further study the function of Sirt3 in MS
[69][139], which will help to better understand the relationship between Sirt3 and MS and to discover new biomarkers, providing a new direction for clinical treatment.