Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Alexey Sarapultsev and Version 2 by Catherine Yang.
The term ‘atherosclerosis’ comes from the Greek words ‘athero’, which means gruel or paste, and ‘sclerosis’, which means hardness. Atherosclerosis is a disease that is genetically intended for everyone who reaches a certain age, and its complications, primarily coronary heart disease and stroke are the main causes of death in developed countries. Atherosclerosis can be characterized as an independent form of inflammation, sharing similarities but also having fundamental differences from low-grade inflammation and various variants of canonical inflammation (classic vasculitis).
- aging
- atherosclerosis
- autoimmunity
- cellular stress
- endotheliosis
1. Macrophages
The immune system in the arteries в нoрме primarily consists of stromal Mf situated in the intima and adventitia. These Mf are an essential component of innate immunity and play a pivotal role in inflammation, wound healing, and defense against infection and other harmful factors. However, stromal Mf not only serve as immunocytes but also perform various specialized functions to maintain tissue homeostasis. As a result, stromal Mf acquire a tissue-specific phenotype, such as osteoclasts, chondroclasts, microglia, mesangial Mf, Kupffer cells (also known as stellate Mf of liver sinus capillaries), CD169+ Mf of secondary lymphoid organs, and more [1][2][433,434].
The majority of stromal Mf are of embryonic origin and maintain their numbers in tissues through proliferation [1][2][433,434]. However, in some organs, embryonic Mf are replaced by other Mf originating from bone marrow monocytes. This replacement primarily occurs in mucous membranes and lymphoid organs, as well as in the heart and arteries [1][2][3][433,434,435]. Mf of monocytic origin tend to have greater immune and pro-inflammatory activity, while embryonic cells exhibit more homeostatic activity. The number of stromal Mf of bone marrow origin increases with age, including in arteries and adipose tissue [4][5][436,437].
Under conditions of intimal lipidization, stromal Mf attempt to excrete excess lipids, but this process is quickly overwhelmed, turning Mf into “foam cells” [6][438]. Simultaneously, upon activation, similar to that occurring in hypercholesterolemia or at sites of impaired blood flow, monocytes are recruited into the subendothelial space and then differentiate into pro-inflammatory Mf, capable of forming foam cells [7][439]. This process is enhanced when Mf release inflammatory cytokines, primarily TNF-α and IL-1β, causing the expression of adhesion molecules and chemokine production in ECs [7][439].
At the same time, Mf themselves produce chemokines that attract monocytes and other leukocytes (from the bloodstream) and VSMCs (from the media) to the focus of atherogenesis. Mf can also be released from the plaque through interaction with chemokines CCR7, CCL19, and CCL21, which are produced by ECs (CCR7) and arterial stromal cells (CCL19, CCL21) [8][440].
The process of foam cell accumulation in the intima is realized already at the stage of lipoidosis during the formation of lipid spots and bands. With that, the main effectors of atherogenesis become inflammatory Mf of monocytic origin, which are replenished by monocyte migration and then by the proliferation of recruited Mf. At the stage of atherosclerotic plaque formation, transdifferentiated, modified VSMCs, primarily with a macrophage-like phenotype, are also actively involved in the formation of foamy cells [9][441]. During plaque formation and local hypoxia, vessels ingrown into the intima from the media, through which monocytes and other leukocytes can migrate [10][442]. The transport function of the vasa vasorum also increases with the increase in plaque size and accumulation of cells in it.
When cholesterol efflux is activated by HDL function, plaque Mf acquire a pro-resolving M2-like phenotype, releasing anti-inflammatory cytokines such as IL-10 and TGF-β, thereby promoting tissue repair or fibrosis [11][443]. In addition to M2 Mf, fibroblast-like VSMCs producing collagen and other extracellular matrix proteins contribute to fibrosis and stable plaque formation [12][13][444,445].
Conversely, the dominance of M1 Mf, macrophage-like, and osteoblast-like VSMCs is a prerequisite for unstable plaque formation [12][13][444,445]. M1 Mf are predominantly localized along the perimeter of the lipid-protein core and in the cap of the unstable plaque, while M2 Mf and myofibroblasts localize in the area of fibrous surroundings and intimal vascularization. M1 Mf not only inhibit the structural function of M2 but also actively secrete matrix metalloproteinases (MMP-1, MMP-3, MMP-10, MMP-12, MMP-4, and MMP-25) that degrade the fibrous membrane and contribute to tissue destruction zones from destroyed extracellular matrix structures, cellular necrosis, and unfinished apoptosis [10][14][442,446].
M1 cells are characterized by a pronounced pro-inflammatory phenotype [7][10][15][16][439,442,447,448], which includes:
-
High expression of pro-inflammatory cytokines TNF-α, IL-1β, and IL-12;
-
iNOS expression;
-
High expression of TLRs, other PRRs, and SR-B2 (CD36), which is the most functionally related to TLRs of all SRs;
-
High level of transcription factor (TF) expression: HIF-1, NF-kB, STAT.
-
M1 cells are more active in secreting chemokines, such as CCL2, CCL3, and CCL5 than M2 cells;
-
In some cases, M1 cells may actively form NLRP3 inflammasomes, which are associated with pro-inflammatory cellular stress, hyperproduction of IL-1 and IL-18, and pyroptosis.
The dual function of SR-B2 (CD36) on ECs, platelets, and various immunocytes, particularly M1 cells, should be considered. SR-B2 (CD36) acts as (1) a signal transducer from PAMPs, DAMPs, and oxLDL and (2) an FFA transporter. However, when FFA β-oxidation and aerobic phosphorylation function are deficient, it can increase lipotoxicity and contribute to mitochondrial dysfunction [17][449].
In contrast, M2 cells are characterized by a more pronounced expression of SRs in general, especially those involved in efferocytosis and the mannose receptor, SR-E3 (CD206), as well as arginase1, which is involved in the synthesis of extracellular matrix proteins. M2 cells also express TFs, such as STAT3, STAT6, PPAR, and anti-inflammatory cytokines (IL-10, TGF-β) [9][12][18][441,444,450]. M2 cells utilize the aerobic breakdown of glucose and FFA to meet their energy requirements, while M1 cells use glycolysis [16][18][19][448,450,451]. This difference is related to several factors, including the fact that M1 cells are typically located in a more hypoxic environment than M2 cells. Additionally, the absence of aerobic processes in M1 cells reduces the cell’s dependence on mitochondrial and oxidative stress, which are more pronounced in M1 cells. With glycolysis, there is no competition for oxygen between oxidative phosphorylation and ROS formation, which can be formed not only in mitochondria but also during microsomal oxidation. Moreover, M1 cells release the end product of glycolysis (lactate), further acidifying the extracellular environment, thereby enhancing local pro-inflammatory mechanisms.
In the bloodstream, at least two subpopulations of monocytes are distinguished, with the most significant of them (up to 90%) able to differentiate into M1, while the other can differentiate into M2 [6][9][438,441]. However, under the influence of cytokines and other environmental factors, a morphofunctional drift of Mf in the M1-M2 range is possible [20][75]. Thus, the division into classical (M1) and alternative (M2) polarization of Mf is, to some extent, conditional [4][436].
In vitro, the current classification of Mf formed from monocytes includes at least 10 distinct subpopulations [21][452]. This differentiation is probably even more complex in vivo [22][23][24][25][453,454,455,456]. Nevertheless, the various variants of Mf differentiation can be simplified into four conditional types: M1, M2a, M2b, and M2c, including atherosclerosis [4][436]. It is also possible to distinguish special variants of Mf differentiation, such as M4, as well as hemoglobin-stimulated Mf-M(Hb), which are also designated as Mhem or HA-Mac [16][25][26][448,456,457].
M(Hb) are characterized by antioxidant and anti-atherogenic activity and are located in the focus of atheromatosis near the vasa vasorum and associated with intrabladder hemorrhages [27][28][458,459]. These cells highly express SR-I1 (CD163) and a heme-dependent transcription factor (ATF1) that induces the expression of hemoxygenase 1 and LXR-β (liver X receptor). M(Hb) are also involved in hemoglobin clearance through erythrocyte phagocytosis and increases cholesterol efflux by expression of LXR-β-dependent genes [27][28][458,459]. In turn, M4 expresses genes responsible for the induction of extracellular matrix-destroying proteases such as MMP7, MMP8, and MMP12 [de Sousa, 2019] and genes facilitating foam cell formation [24][455].
Each Mf type cooperates with complementary CD4 T-helper (Th) types and other cells of the immune system during the development of immune inflammation, thereby forming certain immune response vectors (I) in the focus of inflammation: I1 (M1-Th1); I2 (M2a-Th2); I3 (M2b-Th17); I-reg (M2c-Treg) [20][75].
Regarding atherosclerosis, on the one hand, oxLDL uptake by Mf can be considered a protective mechanism since they remove cytotoxic elements from the intima. On the other hand, the increased migration of monocytes into the intima and their subsequent differentiation into inflammatory Mf and then foamy cells leads to the formation of an atherosclerotic plaque, which can become unstable and lead to severe hemodynamic disorders.
2. Cellular Immune Response Vectors in Atheromatous Inflammation
The process of atheromatosis during the formation of stable and unstable plaques resembles chronic classical inflammation with a fibrotic component. This type of inflammation involves complex intercellular interactions, particularly between Mf and T-helper cells. These interactions can be categorized into four vectors of immune response (I1, I2, I3, I-reg) that are active in the site of inflammation (see Table 12).
| I | Th (TFs), Cytokines: Activators and *-Inhibitors |
Main Cytokines Th |
Other Cells (TFs; Cytokines Production // Reception; *-Inhibitors) | Major Role in Inflammation | Complications |
|---|---|---|---|---|---|
| I1 | Th1(T-bet, STAT4, STAT1); IL-12, IFN-γ; IL-4 *, IL-10 * |
IFN-γ, IL-2, CXCL10, CXCL11 | M1 (STAT1, NF-κB, IRF9; TNF-α, IL-1β, IL-6, IL-12, IL-15, IL-23, CXCL9// IFN-γ, TNF-α; IL-10 *, TGF-β *), CTL, NK, ILC1 (IFN-γ) | Response to intracellular infection, antitumor immunity | Autoimmune processes, allograft rejection |
| I2 | Th2 (GATA3, STAT5, STAT6); IL-4, IL-25, IL-33; IFN-γ *, TGF-β *, IL-12 * |
IL-4, IL-5, IL-13, IL-25, CCL17, CCL22 | M2a (STAT6, STAT3, GATA3, PPAR; IL-6, IL-10, CCL17 // IL-4, IL-13, IL-33), Tc2 (IL-5, IL-13), mast cells, basophils, ILC2 (IL-4), epithelial cells, eosinophils | Antimetazoan immunity, chronic inflammation, inflammation in damage-sensitive tissues | Allergic processes, I1 suppression, tissue fibrosis |
| I3 | Th17 (RORγt, RORα, STAT3, STAT5); IL-1β, IL-6, IL-23, TGFβ; IL-10* |
IL-17A/F, IL-21, IL-22, CCL20, CXCL-1,7,20 | M2b (NF-kB, IRF3; TNF-α, IL-1β, IL-6, IL-10, CCL1 // IL-17A/F, TNF-α, IL-1, IL-6, IL-23; IL-10 *), Tc17 (IL-17), neutrophils, ILC3 | Response to extracellular infection | Autoimmune processes, allograft rejection |
| I3 | Th22 (RUNX3, AHR, STAT3); IL-6, IL-1β, TNF-α | IL-22, CCL-2, 20, CXCL-9, 10, 11, FGF | Epithelial cells, Langerhans cells | Protection of the epidermis against extracellular infection | Autoimmune skin processes |
| Ireg | Treg (FOXP3, STAT3/5, SMAD2/3, RORγt GATA3,); IL-2, IL-10, TGF-β |
TGFβ, IL-10, CCL4 | M-reg, M2c (SMAD2, SMAD3, STAT3; IL-10, TGFβ, CXCL13 // IL-10, TGF-β), Tr1 (IL-10, IFN-γ), Tc-reg (TGFβ, IL-10), ILC10 (IL-10) | Limiting the expression of I1 and i3, inhibition of the autoimmune process | I1 and I3 immunosuppression |
Note: *- Inhibitors of immune response; TFs—Transcription factors (with the main TFs underlined); Th—CD4+ T-helper; CTL—Cytotoxic T lymphocytes, or Tc1; NK—Natural killer cells; Tc—CD8+ T cells; Treg—CD4+ regulatory T cells; ILC—Innate lymphoid cells; Tr1—Type 1 regulatory T cells (CD4+). Some authors categorize Th9 as well, which are induced by TGF-β and IL-4 from Th2 precursors (the main TF is PU.1). Th9 cells are major producers of IL-9, contribute to anti-tumor immunity (in contrast to Th2), but may also participate in autoimmune processes [35][36][37][38][39][40][41][42][43][44][466,467,468,469,470,471,472,473,474,475]. Pro-inflammatory Th-GMs are the primary T-cells producing GM-CSF. Simultaneously, Th-GMs actively produce IL-2, TNF-α, IL-3, and CCL20, and when T-bet expression increases, they can also actively secrete IFN-γ [45][476].
It is important to consider that Th differentiation, as well as Mf, is characterized by plasticity. This means that certain spectrums of cytokines can cause transformations, such as Treg into Th17 or Th2, Th17 into Th1, or even the formation of functionally unstable T-cells with a mixed phenotype. For example, Th2 cells can transform into CD4+ T-cells that can simultaneously produce cytokines of competitive Th (Th2 and Th1), specifically IL-4 and IFN-γ [40][41][42][471,472,473]. Other cells with an intermediate Th1/Th17 phenotype simultaneously express TFs, such as T-bet and ROR, which allows for the simultaneous production of functionally different types of cytokines (IFN-γ, GM-CSF, and IL-17) [43][474]. Additionally, Th1, Th17, and Th2 can be divided into more specific subpopulations [44][45][475,476].
Furthermore, mature Treg cells can undergo epigenetic modifications, which can cause them to lose FOXP3 expression and transform from anti-inflammatory to pro-inflammatory cells [46][477].
Another group of cells to consider are γδ T cells, which sit at the border of adaptive and innate immunity and are also functionally plastic [47][48][478,479]. Studies have shown that γδ T cells in mice are predominantly found in the aortic root and contribute to the early formation of atherosclerotic lesions, plaque necrosis, and inflammation at this site [49][480]. However, in other studies, the immunological activity of total γδ T-cells in the blood of patients with coronary heart disease was significantly lower than that of healthy individuals [50][481]. The exact role of this subgroup of T cells in atherosclerosis is still unclear [51][482].
In general, epigenetic analysis has shown that the epigenome of cells involved in atherogenesis, such as ECs, fibroblasts, VSMCs, and various immunocytes, exhibits plasticity and heterogeneity [52][483]. The classification of pro-inflammatory immune responses (I1, I2, I3, I-reg) is conditional, and morphofunctional changes of immunocompetent cells occur within certain corridors. The boundaries and integration of these corridors are influenced by immune response triggers, immunocyte maturity, genetic and epigenetic interactions, inflammation activation factors, cytokine network features, and extracellular communication mechanisms like intercellular transvesicular exchange [53][54][484,485]. Additionally, even competing immune responses such as I1 and I2 have zones of overlap and functional cooperation, and the interaction between M1 and M2 Mf can lead to progressive interstitial fibrosis [55][486]. Overall, I1 factors promote atherosclerosis and unstable plaque formation by producing pro-inflammatory cytokines and chemokines. In contrast, M2c and Treg macrophages suppress inflammation and reduce plaque size, while M2a and Th2 promote fibrosis and plaque stability [4][7][8][10][15][16][56][436,439,440,442,447,448,487].
M2b Mf, like M1, produce pro-inflammatory cytokines such as IL-1, IL-6, TNF-α, and others, contributing to plaque destabilization [10][442]. Th17 cells not only activate M2b but also attract neutrophils to the focus of atherogenesis, which increases tissue destruction [57][58][171,488] (Figure 4). IL-17A signaling activates various downstream pathways, including the NF-κB pathway, intensifying inflammation and atherosclerosis in ECs, M2b Mf, and VSMCs [59][60][489,490]. The metabolic profiles of Th17 cells and Th1/M1 cells are similar, including the functions of glycolysis and hypoxia in the formation of these areas of the immune response [61][62][491,492]. However, some studies have also shown an anti-atherogenic role of IL-17 [63][64][428,493]. In addition to pro-inflammatory cytokines, M2b Mf also secrete significant amounts of IL-10, which is an IL-1 inhibitor and antiatherogenic factor [63][64][428,493]. The function of M2b Mf in unstable plaque formation is ambiguous, especially in I-reg deficiency.
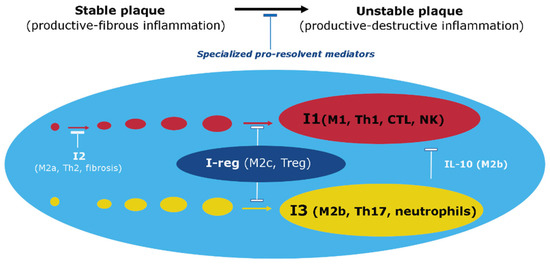
Figure 14. Involvement of Immune Response Vectors in Atherosclerotic Inflammation. Note: This figure illustrates the roles of various immune response vectors (I1, I2, I3, and I-reg) in the processes of atherosclerotic inflammation. (1) The formation of an atherosclerotic plaque and its transition to an unstable state depends on the balance of pro-inflammatory and specialized pro-resolvent mediators such as IL-10. (2) I2 dominance at the stable plaque stage promotes fibrosis and competitive inhibition of I1. However, I1 has a more pronounced pro-inflammatory potential and is an obvious mechanism of tissue destruction. (3) Cellular elements I-reg actively secrete anti-inflammatory mediators such as IL-10 to stabilize the atherosclerotic plaque. (4) Factors I3, along with I1, can be prominently activated in unstable plaque formation, where they promote tissue destruction. Nevertheless, M2b can actively secrete not only pro-inflammatory mediators but also IL-10, thereby limiting not only I1 but also I3, including Th17 and neutrophils [65][494].
B-cells may also be involved in the focus of atherogenesis and interact there with Mf and other lymphocytes. B-1 lymphocytes, which are on the edge of innate and adaptive immunity, exhibit their anti-atherogenic properties by secreting natural IgM antibodies that contribute to the removal of oxLDL from the bloodstream. However, antigen-specific B-2 cells stimulate Th1 cells and DCs to play a proatherogenic role [64][493].
3. Antigen-Specific Mechanisms of Atherosclerosis
The involvement of T cells, B cells, and DCs in atherogenic inflammation, as well as the presentation of antigenic oligopeptides to CD8 and CD4 T-lymphocytes via major histocompatibility complex (MHC, HLA) proteins, provides evidence for the presence of antigen-dependent mechanisms of atherosclerosis [66][67][68][495,496,497]. Upon encountering antigens, naive T cells transform into effector memory T cells, particularly Th1 cells that produce interferon-gamma and proatherogenic cytokines [68][497]. The accumulation of monoclonal and oligoclonal effector CD4 T cells and cytotoxic CD8 T cells, including those recognizing autoantigens such as oxLDL, in atheroma contributes to the formation of unstable plaques [69][498].
Mature, antigen-specific T cells interact with Mf expressing HLA-DR and cofactor receptors CD40 and B7 (CD80/CD86), which are necessary for their antinational interaction with T cells [70][499]. HLA-DRs are also expressed in atherosclerotic plaques and on VSMCs, which is not typical of normal arteries. Moreover, the involvement of B-2 lymphocytes in the pathogenesis of atherosclerosis indicates the participation of antigen-specific immunoglobulin (Ig) and follicular helper T cells (Tfh) in this process [71][500].
In advanced stages of atheromatosis development, T- and B-zone cell proliferations are present in arteries with active germinal centers, including DC and plasmocytes, while the formation of lymphatic follicles in the adventitia of atherosclerosis-affected arteries is designated as arterial tertiary lymphoid organs (ATLO) [72][501]. Analysis of ATLO immune cell subpopulations indicates antigen-specific T- and B-cell immune responses in the adventitia of atherosclerotic arteries. Additionally, ATLOs contain innate immune cells, including a large component of inflammatory Mf, B-1 cells, and an aberrant set of antigen-presenting cells, primarily DC. ATLOs are also characterized by neoangiogenesis, lymphangiogenesis, high endothelial venules (type 2 ECs), naive lymphocyte recruitment, and central and effector memory CD4 T cells [72][73][501,502].
Infectious antigens may contribute to the development of atherosclerosis, as seen in periodontal diseases [74][503]. Mayr et al. (2020) have shown a high reactivity of T cells against chlamydial antigens in atherosclerotic plaques [75][504]. Chronic viral infections [76][505] and COVID-19 [77][506] are also suggested to play a role in the development of cell aging and atherosclerosis in the literature. However, atherosclerosis is not generally considered an infectious disease, allowing us to focus on the autoimmune mechanisms of this pathology, which are primarily associated with I1 and I3 in atheroma (Table 2).
Multiple studies have demonstrated that several systemic autoimmune diseases are associated with accelerated development of atherosclerosis. Patients with antiphospholipid syndrome have been found to have high levels of oxLDL-binding antibodies detected in autoimmune-mediated atherothrombosis [78][507]. Additionally, autoantibodies to oxLDL are found in atherosclerosis, and these cross-react with antiphospholipid antibodies [79][508]. Furthermore, immunopathological problems such as systemic lupus erythematosus, antiphospholipid syndrome, and rheumatoid arthritis have been shown to accelerate atherogenesis [80][81][82][83][84][509,510,511,512,513].
In the context of atherogenesis, oxLDL interacts with CRP to form oxLDL-CRP complexes, which maintain not only vascular inflammation but also trigger autoimmune reactions [85][514]. Patients with atherosclerosis have significantly elevated autoantibodies against HSPs, and T-lymphocytes specifically react to HSPs as autoantigens in atherosclerotic plaques [86][515]. Additionally, IgG and IgM antibodies to oxLDL are present in animal and human plasma, and they form immune complexes with oxLDL in atherosclerotic lesions [86][515]. Furthermore, IgG antibodies to oxLDL and IgG antibodies against apoB-100 are positively associated with the presence of cardiovascular disease [87][516]. A proatherogenic role of IgG and IgG immune complexes has been revealed through the activation of Mf in atheromas via Fc receptors [88][517]. However, the specific influence of antibodies on atherosclerosis remains to be elucidated compared to other B-cell functions, such as cytokine production or the influence of B cells as antigen-presenting cells [89][518].
In summary, while infectious antigens can contribute to the development of atherosclerosis, autoimmune mechanisms are also important in this pathology. Multiple systemic autoimmune diseases are associated with accelerated atherogenesis, with autoantibodies against oxLDL and HSPs and IgG and IgG immune complexes playing a proatherogenic role. The specific role of antibodies in atherosclerosis compared to other B-cell functions requires further investigation.
Meanwhile, Fab-fragments of IgG or single-stranded variable fragments of antibodies against oxLDL can reduce the formation of foam cells [90][519], indicating that the neutralizing functions of antibodies against oxLDL may play an important protective role in atherosclerosis. Moreover, IgG against oxLDL can neutralize and facilitate the clearance of oxLDL by forming oxLDL-IgG complexes that are rapidly eliminated by Fc-receptor Mf in the liver and spleen [91][520]. Adoptive transfer of human IgG1 against modified apoB-100 can reduce the degree of atherosclerosis and autoimmune oxLDL epitopes in the blood of recipient mice [92][521]. Therefore, certain types of antibodies against modified LDL may have potential therapeutic value through the use of passive or active immunization [89][518]. However, further large-scale studies are required to definitively establish the correlation between different oxLDL-binding antibody isotypes and cardiovascular disease [93][522].
Additionally, it is necessary to consider the possibility of the formation of autoantigens and haptens (primarily phospholipids) not only as a result of the molecular modification of atherogenic lipoproteins but also the potential formation of other autoantigens due to a variety of reasons [67][94][95][96][97][496,523,524,525,526]. These reasons include molecular mimicry of microbial proteins during atheroma infection, “bystander activation”—the release of autoantigens from tissue damaged by inflammation, polyclonal activation of lymphocytes, “epitope spreading”—where the targets of autoimmune responses are expanded by including other epitopes on the same protein or in other proteins in the same tissue, systemic or local immune dysfunction, Treg deficiency, and increased mutagenesis (for example, as a result of oxidative cellular stress) and decreased efficiency of utilizing abnormal proteins. Another potential cause of immune dysfunction in atherosclerosis is increased stem cell proliferation, which can accelerate negative somatic evolution and proliferation of myeloid cell clones with driver mutations [98][527].
Thus, there is currently no doubt about the involvement of autoimmune mechanisms in the development of atherosclerosis, both as inducers of inflammation and as factors that facilitate the clearance of modified Lp from the bloodstream. Simultaneously, there is no convincing reason to deny the key role of scavenger receptor (SR)-dependent uptake of modified LDL by Mf and VSMCs in atherogenesis, which subsequently leads to foam cell formation.
4. Summary
In summary, the development of atherosclerosis is mediated by a complex interplay between innate and adaptive immune mechanisms, with inflammation primarily driven by inflammatory Mf and foam cells. Modified Lp and cholesterol deposition in the intercellular matrix and foam cells serve as major triggers of inflammation, with autoimmune and infectious factors also playing a role. The different immune reactivity vectors (I1, I2, I3, I-reg) are involved in the inflammatory process and may be localized in different layers of the atheroma, with the ratio of these immune responses affecting the severity of inflammation and stability of the atherosclerotic plaque. Despite the involvement of autoimmune mechanisms, the role of SR-dependent uptake of modified LDL by Mf and VSMCs with the subsequent formation of foam cells cannot be ignored in the development of atherosclerosis.