RAS
genes encode signaling proteins, which, in mammalian cells, act as molecular switches regulating critical cellular processes as proliferation, growth, differentiation, survival, motility, and metabolism in response to specific stimuli. Deregulation of Ras functions has a high impact on human health: gain-of-function point mutations in
RAS
genes are found in some developmental disorders and thirty percent of all human cancers, including the deadliest. For this reason, the pathogenic Ras variants represent important clinical targets against which to develop novel, effective, and possibly selective pharmacological inhibitors. Few druggable sites have been identified for wild type and some oncogenic Ras mutants, and few natural compounds able to attenuate Ras signaling have been identified so far. Natural products represent a virtually unlimited resource of structurally different compounds from which one could draw on for this purpose, given the improvements in the isolation and screening of active molecules from complex sources, which can now be exploited for the selection of potential Ras inhibitors from natural sources.
- Ras signaling
- cancer
- Ras inhibitory strategies
- Ras druggable pockets
- natural products
- anticarcinogenic effect
1. Introduction
1.1. Ras Pproteins
Ras proteins are eukaryotic small guanine nucleotide-binding (G) proteins that, by cycling between the GDP-bound inactive state and the GTP-bound active state, act as molecular switches in signaling pathways regulating many cellular processes including proliferation, growth, survival, adhesion, migration and metabolism in mammalian cells [1]. Ras proteins are endowed with low intrinsic GTPase activity and a very slow rate of spontaneous nucleotide exchange. The Ras activation state is finely regulated, in response to different specific extracellular stimuli, by the competitive interplay of upstream regulators: Guanine nucleotide Exchange Factors (GEFs) and GTPase Activating Proteins (GAPs). GEFs activate Ras proteins by promoting nucleotide dissociation, and thereby preferentially GDP/GTP exchange due to GTP being 10-fold more abundant in cells than GDP, while GAPs inactivate them by providing an essential catalytic group for GTP hydrolysis [2][3][4][5]. In the active GTP-bound state Ras proteins increase their affinity for many effectors that initiate downstream signal transduction [6][7] (Figure1). In human cells, three
Ras proteins are eukaryotic small guanine nucleotide-binding (G) proteins that, by cycling between the GDP-bound inactive state and the GTP-bound active state, act as molecular switches in signaling pathways regulating many cellular processes including proliferation, growth, survival, adhesion, migration and metabolism in mammalian cells [1]. Ras proteins are endowed with low intrinsic GTPase activity and a very slow rate of spontaneous nucleotide exchange. The Ras activation state is finely regulated, in response to different specific extracellular stimuli, by the competitive interplay of upstream regulators: Guanine nucleotide Exchange Factors (GEFs) and GTPase Activating Proteins (GAPs). GEFs activate Ras proteins by promoting nucleotide dissociation, and thereby preferentially GDP/GTP exchange due to GTP being 10-fold more abundant in cells than GDP, while GAPs inactivate them by providing an essential catalytic group for GTP hydrolysis [2–5]. In the active GTP-bound state Ras proteins increase their affinity for many effectors that initiate downstream signal transduction [6,7] (Figure1). In human cells, three
RAS
genes (
HRAS
,
NRAS
and
KRAS
), encode four homologous but functionally distinct isoforms, HRas, NRas, KRas4A and KRas4B, the two latter ones deriving from alterative splicing of the
KRAS gene [8][9]. Notably KRas4B is usually referred as KRas.The four isoforms share 90% of sequence identity in the first 166 residues and mainly differ in the carboxyl-terminal hypervariable region (HVR) that contains sites for posttranslational modifications (PTM). This region is responsible for membrane tethering of Ras proteins that is required for correct membrane trafficking and localization, and function of each isoform [10][11].
gene [8,9]. Notably KRas4B is usually referred as KRas.The four isoforms share 90% of sequence identity in the first 166 residues and mainly differ in the carboxyl-terminal hypervariable region (HVR) that contains sites for posttranslational modifications (PTM). This region is responsible for membrane tethering of Ras proteins that is required for correct membrane trafficking and localization, and function of each isoform [10,11].
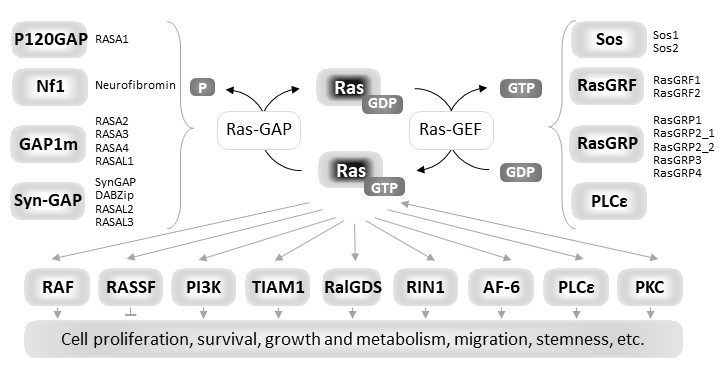
Figure 1.
Diagrammatic representation of the functional cycle, upstream regulators and downstream effectors of Ras proteins
1.2. Ras mutants in Hhuman Ddiseases
Deregulation of Ras activity has a driving role in the pathogenesis of several human diseases, including developmental disorders, known as RASopathies [12][13], and thirty percent of all human cancers, including the deadliest.
Deregulation of Ras activity has a driving role in the pathogenesis of several human diseases, including developmental disorders, known as RASopathies [12,13], and thirty percent of all human cancers, including the deadliest.
KRAS
gene is the most frequently mutated isoform (21%), followed by mutations in
NRAS
(8%), and in
HRAS
(3%) (
www.sanger.ac.uk/genetics/CGP/cosmic/). A mutationally activated KRas oncoprotein is present in almost all pancreatic ductal adenocarcinomas and in up to 50% colorectal cancers. The large majority of gain-of-function missense mutations that constitutively activate Ras oncoproteins map at codons 12 (89%), 13 (9%), and 61 (1%) [14], which encode critical residues involved in the interplay between Ras, nucleotides, and modulators. Each oncogenic mutation alters the functional cycle of Ras through a distinct mechanism depending on the conformational change induced by the presence of the mutated amino acid. For example, the G12V substitution abolishes the intrinsic and GAP-mediated GTP hydrolysis due to interference with the allosteric switch [15][16], while G13D mutation determines the self-sufficiency in nucleotide dissociation, even maintaining the sensitivity to GEFs and at least one GAP [15][16][17][18][19][20]. The Q61L mutation reduces the intrinsic, in both free and Raf-bound Ras, and GAP-mediated GTP hydrolysis and accelerates nucleotide exchange [16]. Regardless of the activation mechanism, all oncogenic Ras mutants show an altered residence time in the GTP-bound active state [15], and aberrantly transduce downstream signals contributing to tumor onset, maintenance, and progression [21], impinging on most cancer hallmarks [22], such as growth signal-independent sustained proliferation, resistance to apoptosis, the ability to migrate and to invade/metastasize, the ability to promote angiogenesis, and ability to elude the immune response, as previously reviewed [23]. Oncogenic
). A mutationally activated KRas oncoprotein is present in almost all pancreatic ductal adenocarcinomas and in up to 50% colorectal cancers. The large majority of gain-of-function missense mutations that constitutively activate Ras oncoproteins map at codons 12 (89%), 13 (9%), and 61 (1%) [14], which encode critical residues involved in the interplay between Ras, nucleotides, and modulators. Each oncogenic mutation alters the functional cycle of Ras through a distinct mechanism depending on the conformational change induced by the presence of the mutated amino acid. For example, the G12V substitution abolishes the intrinsic and GAP-mediated GTP hydrolysis due to interference with the allosteric switch [15,16], while G13D mutation determines the self-sufficiency in nucleotide dissociation, even maintaining the sensitivity to GEFs and at least one GAP [15–20]. The Q61L mutation reduces the intrinsic, in both free and Raf-bound Ras, and GAP-mediated GTP hydrolysis and accelerates nucleotide exchange [16]. Regardless of the activation mechanism, all oncogenic Ras mutants show an altered residence time in the GTP-bound active state [15], and aberrantly transduce downstream signals contributing to tumor onset, maintenance, and progression [21], impinging on most cancer hallmarks [22], such as growth signal-independent sustained proliferation, resistance to apoptosis, the ability to migrate and to invade/metastasize, the ability to promote angiogenesis, and ability to elude the immune response, as previously reviewed [23]. Oncogenic
KRAS activation also induces significant changes in cell metabolism, including enhancement in glucose transport and aerobic glycolysis [24][25][26] that determine the acquisition of the hyperglycolytic phenotype known as the Warburg effect [27][28], anaplerotic usage of glutamine [29][30][31], altered sulfur amino acid metabolism [32], altered mitochondrial morphology and function, and production of large amounts of reactive oxygen species (ROS) [33][34]. Ras GAPs and members of the RASSF family constitute a barrier to Ras-dependent transformation in cells. However most Ras oncoproteins are insensitive to GAP, and loss of function of Ras GAPs or RASSFs is common in tumors [35].
activation also induces significant changes in cell metabolism, including enhancement in glucose transport and aerobic glycolysis [24–26] that determine the acquisition of the hyperglycolytic phenotype known as the Warburg effect [27,28], anaplerotic usage of glutamine [29–31], altered sulfur amino acid metabolism [32], altered mitochondrial morphology and function, and production of large amounts of reactive oxygen species (ROS) [33,34]. Ras GAPs and members of the RASSF family constitute a barrier to Ras-dependent transformation in cells. However most Ras oncoproteins are insensitive to GAP, and loss of function of Ras GAPs or RASSFs is common in tumors [35].
2. Natural Products Ttargeting Bbiosynthesis, Pprocessing, Aactivity, and Ssignaling of Ras Ooncoproteins
Due to the critical role of Ras oncoproteins in cancer, many efforts, mostly promoted by the RAS initiative (https://www.cancer.gov/research/key-initiatives/ras), have been devoted to explore different direct and indirect strategies for attenuating their aberrant signaling, as recapitulated in several recent reviews [36][37][38], and schematically depicted in Figure 2. Bioactive natural products identified in some of these strategies are reported in the figure.
Due to the critical role of Ras oncoproteins in cancer, many efforts, mostly promoted by the RAS initiative (https://www.cancer.gov/research/key-initiatives/ras), have been devoted to explore different direct and indirect strategies for attenuating their aberrant signaling, as recapitulated in several recent reviews [36–38], and schematically depicted in Figure 2. Bioactive natural products identified in some of these strategies are reported in the figure.
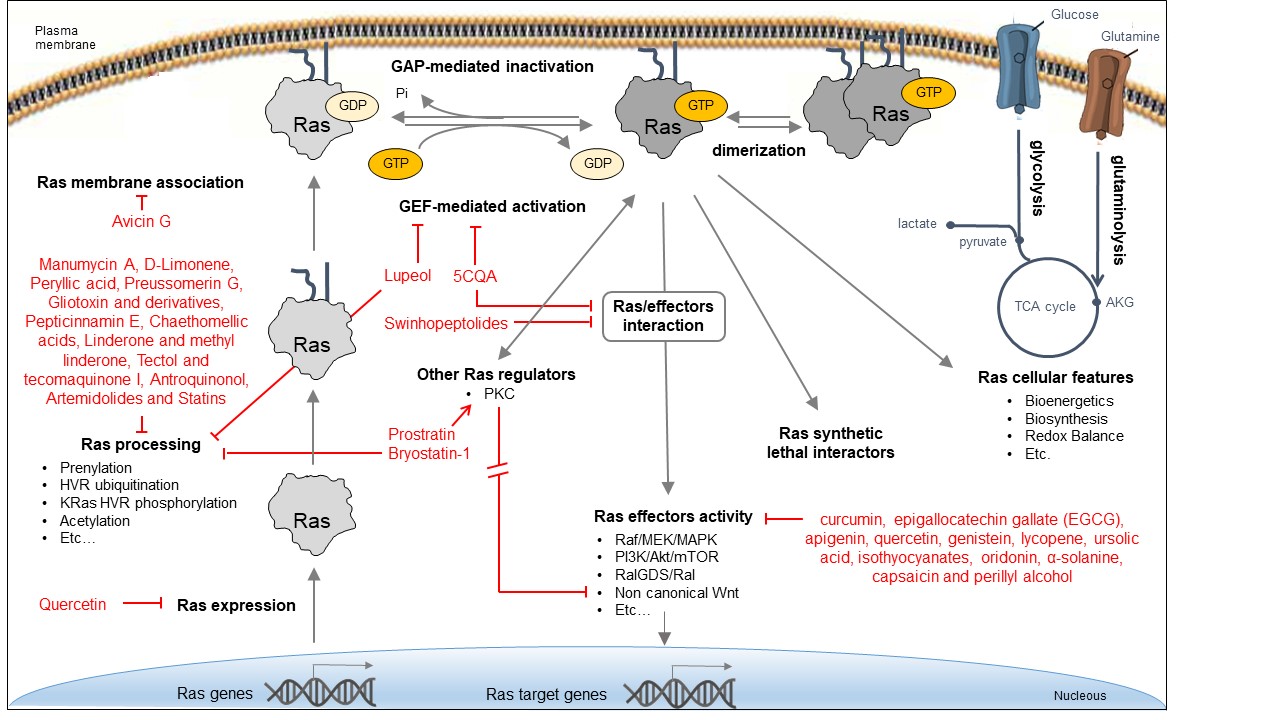
Figure 2.
Approaches for inhibiting Ras oncoproteins biosynthesis, processing, activity, and signaling. Indirect Ras inhibitory approaches include the interference with different processes: expression of Ras oncogenes; Ras processing and membrane localization; activity of Ras regulators; activity of downstream effectors; Ras-dependent cellular features; and activity of synthetic lethal interactors. Direct Ras inhibitory approaches include the interference with Ras/GEF interaction and exchange activity, Ras/effector interaction, and Ras dimerization.
2.1. NPs Indirectly Targeting Ras Function
2.1. NPs indirectly targeting Ras function
2.1.1. NP Inhibiting Ras Expression
2.1.1. NP inhibiting Ras expression
- Quercetin
Quercetin is a dietary flavonoid found in tea, onions, grapes, wines, and apples, and the anti-cancer activities of this compound have been previously explored in breast and colon cancer cells [39]. Quercetin reduced the expression of numerous prostate tumor‑associated microRNAs (miRs) [40]. Quercetin regulated cisplatin sensitivity of human osteosarcoma cells by modulating the miR-217-
KRAS
axis [41]. Consistently, quercetin reduced the steady state levels of K-, H-, and N-Ras mRNAs and proteins in both colon cancer cell lines and primary colorectal tumors [42].
2.1.2. NPs Inhibiting Ras Regulation and Membrane Association
2.1.2. NPs inhibiting Ras regulation and membrane association
- Avicin G
Avicin G is a family of natural plant-derived triterpenoid saponins from
Acacia victoriae
, which mislocalizes KRas from the plasma membrane and disrupts plasma membrane spatial organization of KRas and HRas oncoproteins by depleting phosphatidylserine and cholesterol contents, respectively, at the inner plasma membrane leaflet [43]. Avicin G inhibits oncogenic K- and H-Ras signal output and the growth of KRas-addicted pancreatic and non-small cell lung cancer cells. Avicin G also perturbs lysosomal activity and disrupts cellular localization and activity of sphingomyelinases, resulting in altered cellular sphingomyelin levels and distribution.
- Bryostatin-1
Bryostatin-1 is a cyclic macrolide isolated from the marine bryozoan
Bugula neritina that acts as a protein kinase C (PKC) agonist, activating PKC isozymes at nanomolar concentrations [44][45]. PKC-mediated phosphorylation of the C-terminal segment of KRas regulates its association with the plasma membrane. In particular, bryostatin-1 induces a rapid translocation of KRas to intracellular membranes such as endoplasmic reticulum (ER) and Golgi apparatus, but also to the outer mitochondrial membrane where K-Ras stimulates apoptosis [46]. Bryostatin 1 is in clinical development as an anti-leukaemic agent and is also in Phase II clinical trials against melanomas, lymphomas, and renal cancer [47].
that acts as a protein kinase C (PKC) agonist, activating PKC isozymes at nanomolar concentrations [44,45]. PKC-mediated phosphorylation of the C-terminal segment of KRas regulates its association with the plasma membrane. In particular, bryostatin-1 induces a rapid translocation of KRas to intracellular membranes such as endoplasmic reticulum (ER) and Golgi apparatus, but also to the outer mitochondrial membrane where K-Ras stimulates apoptosis [46]. Bryostatin 1 is in clinical development as an anti-leukaemic agent and is also in Phase II clinical trials against melanomas, lymphomas, and renal cancer [47].
- Prostratin
Prostratin is a phorbol ester found in the bark of the mamala tree of Samoa,
Homalanthus nutans
(Euphorbiaceae), acting as an activator of atypical PKCs. It can efficiently reduce the interaction of KRas and CaM, rewire non canonical Wnt/Ca
2+
signaling, and suppress malignancy mediated by oncogenic KRas in pancreatic cancers [48].
2.1.3. NPs Targeting Ras Processing
2.1.3. NPs targeting Ras processing
- Manumycin A
Manumycin A is a natural macrolide antibiotic isolated from
Streptomyces parvulus that acts as a potent peptidomimetic inhibitor of Ras farnesylation [49][50][51][52]. Manumycin A significantly inhibits the proliferation and migration of vascular smooth muscle cells (VSMCs), reduces the amount of Ras protein localized at the cytoplasmic membrane, inhibits the phosphorylation of MAPK, and disorganizes the actin fibers [53]. In addition, manumycin A decreases exosome biogenesis in prostate cancer cells and in myofibroblasts primarily via targeted inhibition of the Ras/Raf/ERK1/2 signaling [54][55].
that acts as a potent peptidomimetic inhibitor of Ras farnesylation [49–52]. Manumycin A significantly inhibits the proliferation and migration of vascular smooth muscle cells (VSMCs), reduces the amount of Ras protein localized at the cytoplasmic membrane, inhibits the phosphorylation of MAPK, and disorganizes the actin fibers [53]. In addition, manumycin A decreases exosome biogenesis in prostate cancer cells and in myofibroblasts primarily via targeted inhibition of the Ras/Raf/ERK1/2 signaling [54,55].
- D-Limonene and peryllic acid
D-Limonene is a common monoterpene, found in essential oils of orange, lemon, mandarin, lime, grapefruit and many other plants, with antiproliferative, apoptosis-inducing and chemopreventive effects and, as similar monoterpenes, inhibits Ras prenylation [56][57][58]. The related compound peryllic acid is able to inhibit Ras prenylation targeting both farnesyl transferase (FTase) and geranylgeranyl transferase (GGTase) [59].
D-Limonene is a common monoterpene, found in essential oils of orange, lemon, mandarin, lime, grapefruit and many other plants, with antiproliferative, apoptosis-inducing and chemopreventive effects and, as similar monoterpenes, inhibits Ras prenylation [56–58]. The related compound peryllic acid is able to inhibit Ras prenylation targeting both farnesyl transferase (FTase) and geranylgeranyl transferase (GGTase) [59].
- Preussomerin G
The preussomerins and deoxypreussomerins are phenolic fungal metabolites extracted from the coprophilous fungus
Preussia isomera
and the endophytic fungus
Harmonema dematioides with FTase and GGTase inhibitory properties [60][61][62][63]. Low toxicity synthetic esters derived from these compounds require reductive activation specifically at the cancer cells, resulting from hypoxia and overexpression of reductases. The anticancer activity was determined in cancer cell lines with reported reductase activity such as BC-1 cells and NCI-H187 [64].
with FTase and GGTase inhibitory properties [60–63]. Low toxicity synthetic esters derived from these compounds require reductive activation specifically at the cancer cells, resulting from hypoxia and overexpression of reductases. The anticancer activity was determined in cancer cell lines with reported reductase activity such as BC-1 cells and NCI-H187 [64].
- Gliotoxin and derivatives
Gliotoxin is a sulfur-containing mycotoxin, produced by various pathogenic fungi, including
Aspergillus fumigatus, that inhibits Ras farnesylation and cell growth [49][50]. Some derivatives were developed as GGTase specific inhibitors [65].
, that inhibits Ras farnesylation and cell growth [49,50]. Some derivatives were developed as GGTase specific inhibitors [65].
- Pepticinnamin E
The natural product pepticinnamin E was reported to inhibit protein farnesyl transferases and cell proliferation almost 30 years ago [66][67]. Pepticinnamin E contains a rare N‐terminal cinnamoyl moiety as well as several nonproteinogenic amino acids, which mimics the two substrates of FTase, CAAX, and FPP. Its biosynthetic pathway has only recently been characterized due to loss of the original producer organism [68]. A library of 51 analogues was generated from pepticinnamin E and screened for FTase inhibitory activity [69].
The natural product pepticinnamin E was reported to inhibit protein farnesyl transferases and cell proliferation almost 30 years ago [66,67]. Pepticinnamin E contains a rare N‐terminal cinnamoyl moiety as well as several nonproteinogenic amino acids, which mimics the two substrates of FTase, CAAX, and FPP. Its biosynthetic pathway has only recently been characterized due to loss of the original producer organism [68]. A library of 51 analogues was generated from pepticinnamin E and screened for FTase inhibitory activity [69].
- Chaethomellic acids
Chaetomellic acids are a class of alkyl dicarboxylic acids, isolated from
Chaetomella acutiseta. They are potent and highly specific farnesyl-pyrophosphate (FPP) mimic inhibitors of Ras FTase with lower specificity for GGTases [70][71]. Long-term treatment with chaethomellic acid A can attenuate Ras-dependent progression of renal fibrosis in a murine model of chronic kidney diseases [72].
. They are potent and highly specific farnesyl-pyrophosphate (FPP) mimic inhibitors of Ras FTase with lower specificity for GGTases [70,71]. Long-term treatment with chaethomellic acid A can attenuate Ras-dependent progression of renal fibrosis in a murine model of chronic kidney diseases [72].
- Linderone and methyl linderone
The cyclopentenediones linderone and methyl linderone isolated from the fruits of
Lindera erythrocarpa (Lauraceae) showed FTase inhibitory and anti-tumor activity [73][74].
(Lauraceae) showed FTase inhibitory and anti-tumor activity [73,74].
- Tectol and tecomaquinone I
Tectol and the related compound tecomaquinone I were isolated in a screening for FTase inhibitors; tectol also exhibited significant activity against human leukemia cell lines HL60 and CEM [75][76].
Tectol and the related compound tecomaquinone I were isolated in a screening for FTase inhibitors; tectol also exhibited significant activity against human leukemia cell lines HL60 and CEM [75,76].
- Antroquinonol
A compound with anti-inflammatory activities extracted from the mycelium of
Antrodia camphorate antroquinonol has been shown to exert anticancer effects in lung cancer, liver cancer, and leukemia by inhibiting the activity of both Ras FTase and GGTase [77][78][79].
antroquinonol has been shown to exert anticancer effects in lung cancer, liver cancer, and leukemia by inhibiting the activity of both Ras FTase and GGTase [77–79].
- Artemidolides
Arteminolides (A-D) are dimeric sesquiterpene lactones isolated from
Artemisia spp. with inhibitory activity on FTase [80][81]. These compounds and other similar sesquiterpene lactones from
with inhibitory activity on FTase [80,81]. These compounds and other similar sesquiterpene lactones from
Artemisia inhibited tumor cell growth in a dose-dependent manner [82][83]. In particular, arteminolide C blocked in vivo growth of human colon and lung tumor xenograft [84].
inhibited tumor cell growth in a dose-dependent manner [82,83]. In particular, arteminolide C blocked in vivo growth of human colon and lung tumor xenograft [84].
- Statins
Several statins, comprising natural ones (lovastatin, simvastatin), efficiently inhibited KRas protein trafficking from the cytoplasm to the cell membrane of pancreatic cancer cells due to depletion of the mevalonate pathway's intermediates [85].
Several statins, comprising natural ones (lovastatin, simvastatin), efficiently inhibited KRas protein trafficking from the cytoplasm to the cell membrane of pancreatic cancer cells due to depletion of the mevalonate pathway’s intermediates [85].
2.2. NPs Inhibiting Ras Effectors
2.2. NPs inhibiting Ras effectors
- Several compounds
Many Ras effectors play a relevant role in the onset and progression of Ras-dependent disorders and therefore represent attractive therapeutic targets for drug development. Several inhibitors of Ras-ERK signaling have been developed, including Raf inhibitors and MEK inhibitors, as reviewed [86]. Several natural products were reported to inhibit ERK signaling although the mechanisms of action are often unclear. Among these, we can enlist sulphoraphane, epigallocatechin gallate (EGCG), isothyocyanates, genistein, and perillyl alcohol, (see [87] for a review). Also the inhibition of PI3K-AKT-mTOR pathway has been widely experimented, even with natural products such as lycopene, curcumin, resveratrol, genistein, apigenin, oridonin, α-solanine, and capsaicin [88][89][90][91][92].
Many Ras effectors play a relevant role in the onset and progression of Ras-dependent disorders and therefore represent attractive therapeutic targets for drug development. Several inhibitors of Ras-ERK signaling have been developed, including Raf inhibitors and MEK inhibitors, as reviewed [86]. Several natural products were reported to inhibit ERK signaling although the mechanisms of action are often unclear. Among these, we can enlist sulphoraphane, epigallocatechin gallate (EGCG), isothyocyanates, genistein, and perillyl alcohol, (see [87] for a review). Also the inhibition of PI3K-AKT-mTOR pathway has been widely experimented, even with natural products such as lycopene, curcumin, resveratrol, genistein, apigenin, oridonin, α-solanine, and capsaicin [88–92].
2.3. NPs Targeting Ras Activity Directly
2.3. NPs targeting Ras activity directly
- 5-O-caffeoylquinic acid (5-CQA)
The first natural compound reported to directly target Ras activity was a chlorogenic acid and was identified on the basis of its structural resemblance to previously identified synthetic Ras inhibitors [93][94]. The chlorogenic acids (CGAs) occur ubiquitously in food, representing the most abundant polyphenols in the human diet. Particularly high levels of chlorogenic acid (5-O-caffeoylquinic acid, 5-CQA) were found in coffee beans used to prepare green coffee and, after roasting, black coffee, a widespread drink worldwide. A number of CGA beneficial biological effects, including anti-inflammatory activity, anti-carcinogenic activity, and protection against neurodegenerative diseases were reported. Its mechanism of action is based on the inhibition, upon direct binding to the target, of Ras interaction with both activators and effectors. In addition, viability and MAPKs activation/phosphorylation assays performed on KRas
The first natural compound reported to directly target Ras activity was a chlorogenic acid and was identified on the basis of its structural resemblance to previously identified synthetic Ras inhibitors [93,94]. The chlorogenic acids (CGAs) occur ubiquitously in food, representing the most abundant polyphenols in the human diet. Particularly high levels of chlorogenic acid (5-O-caffeoylquinic acid, 5-CQA) were found in coffee beans used to prepare green coffee and, after roasting, black coffee, a widespread drink worldwide. A number of CGA beneficial biological effects, including anti-inflammatory activity, anti-carcinogenic activity, and protection against neurodegenerative diseases were reported. Its mechanism of action is based on the inhibition, upon direct binding to the target, of Ras interaction with both activators and effectors. In addition, viability and MAPKs activation/phosphorylation assays performed on KRas
G13D
expressing breast cancer cells, MDA-MB-231, suggested its capability of reducing cancer cells growth [95].
- Lupeol
The triterpenoid lupeol was reported to inhibit farnesyl transferase [96] and thus to inhibit the growth of KRas mutant cancer cell lines but not of wild‐type KRas‐expressing cells [97]. Lupeol was identified as a KRas directly binding compound in an in silico screening of a library of the triterpenoid class of molecules and its binding results in inhibition of GDP/GTP exchange [97].
- Swinhopeptolides
Two new cyclic depsipeptides named swinhopeptolides A and B have been isolated from the marine sponge
Theonella swinhoei cf. verrucosa, collected from Papua, New Guinea. These compounds contain 11 diverse amino acids and 13-carbon polyketide moieties attached at the N-terminus. They can impede the interaction between Ras and Raf, a serine/threonine protein kinase. Swinhopeptolides A and B showed significant inhibition of the Ras/Raf signaling pathway with effectiveness in the micromolar range [98].
, collected from Papua, New Guinea. These compounds contain 11 diverse amino acids and 13-carbon polyketide moieties attached at the N-terminus. They can impede the interaction between Ras and Raf, a serine/threonine protein kinase. Swinhopeptolides A and B showed significant inhibition of the Ras/Raf signaling pathway with effectiveness in the micromolar range [98].
3. Conclusion and Pperspectives: Nnatural Pproducts as a Ssource of Sselective Iinhibitors of Ras Ooncoproteins
Although various strategies to inhibit Ras have been explored over three decades, success has only recently been achieved in human clinical trials, in particular with small molecules capable of directly inhibiting the activity of specific Ras oncoproteins, which, although important, represent only a small percentage of those involved in human pathologies. Therefore, there is a strong need for effective inhibitors which target the other pathogenic Ras variants. Several natural compounds has been successfully applied to different direct and indirect strategies of Ras inhibition, however natural product research has not been prioritized in the research and development of Ras inhibitors so far. Since natural products provide a virtually limitless source of structurally novel, highly diverse natural compounds, they would be a promising approach to discover novel molecules with higher affinity for specific pathogenic Ras mutants. Three points are worth mentioning on this subject:
-
the improvement of techniques that allow to isolate, purify and structurally characterize new molecules of natural origin, often already available in large libraries [99][100];[99,100];
the simultaneous development of experimental and computational approaches for their high-throughput screening (HTS) on targets of clinical relevance;
the availability of virtual screening allowing to identify the structurally most promising compounds for a target of interest, thereby reducing the research costs.
Both structure-based virtual screenings and HTS approaches with Ras oncoproteins as targets will now be able to take advantage of the newly-discovered druggable pockets available in specific oncogenic Ras isoforms and mutant proteins to isolate, characterize and iteratively improve Ras-specific inhibitors (Figure 3).
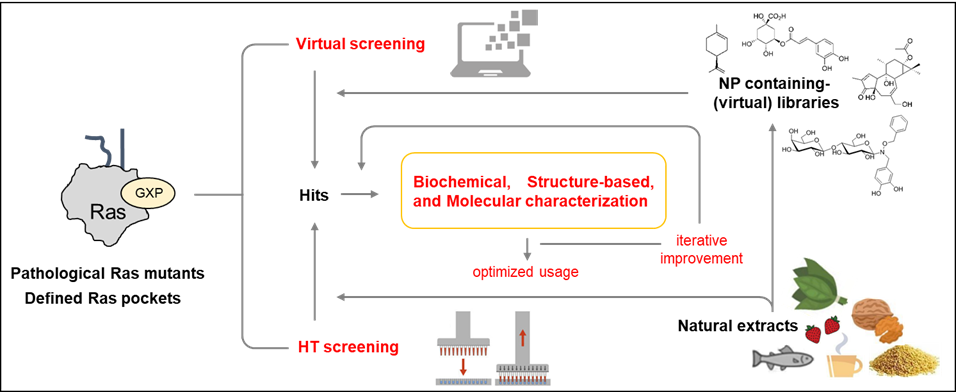
Figure 3. Approaches for the identification and development of Ras inhibitors from natural sources.
. Approaches for the identification and development of Ras inhibitors from natural sources.
References
- Simanshu, D.K.; Nissley, D. V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33, doi:10.1016/j.cell.2017.06.009.
- Boriack-Sjodin, P.A.; Margarit, S.M.; Bar-Sagi, D.; Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343, doi:10.1038/28548.
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmüller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic ras mutants. Science (80-. ). 1997, 277, 333–338, doi:10.1126/science.277.5324.333.
- Ahmadian, M.R.; Stege, P.; Scheffzek, K.; Wittinghofer, A. Confirmation of the arginine-finger hypothesis for the GAP-stimulated GTP-hydrolysis reaction of Ras. Nat. Struct. Biol. 1997, 4, 686–689, doi:10.1038/nsb0997-686.
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell 2007.
- Marshall, C.J. Ras effectors. Curr. Opin. Cell Biol. 1996, 8, 197–204, doi:10.1016/S0955-0674(96)80066-4.
- Nakhaeizadeh, H.; Amin, E.; Nakhaei-Rad, S.; Dvorsky, R.; Ahmadian, M.R. The RAS-effector interface: Isoform-specific differences in the effector binding regions. PLoS One 2016, 11, 1–20, doi:10.1371/journal.pone.0167145.
- Omerovic, J.; Laude, A.J.; Prior, I.A. Ras proteins: Paradigms for compartmentalised and isoform-specific signalling. Cell. Mol. Life Sci. 2007, 64, 2575–2589, doi:10.1007/s00018-007-7133-8.
- Nussinov, R.; Tsai, C.J.; Chakrabarti, M.; Jang, H. A new view of ras isoforms in cancers. Cancer Res. 2016, 76, 18–23, doi:10.1158/0008-5472.CAN-15-1536.
- Ahearn, I.; Zhou, M.; Philips, M.R. Posttranslational Modifications of RAS Proteins. Cold Spring Harb. Perspect. Med. 2018, 8.
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting RAS membrane association: Back to the future for anti-RAS drug discovery? Clin. Cancer Res. 2015, 21, doi:10.1158/1078-0432.CCR-14-3214.
- Bentires-Alj, M.; Kontaridis, M.I.; Neel, B.G. Stops along the RAS pathway in human genetic disease. Nat. Med. 2006, 12.
- Tidyman, W.E.; Rauen, K.A. Pathogenetics of the RASopathies. Hum. Mol. Genet. 2016, 25.
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of ras mutations in cancer. Cancer Res. 2012, 72.
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol. Cancer Res. 2015, 13, 1325–1335, doi:10.1158/1541-7786.MCR-15-0203.
- Smith, M.J.; Neel, B.G.; Ikura, M. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 4574–4579, doi:10.1073/pnas.1218173110.
- Palmioli, A.; Sacco, E.; Airoldi, C.; Di Nicolantonio, F.; D’Urzo, A.; Shirasawa, S.; Sasazuki, T.; Di Domizio, A.; De Gioia, L.; Martegani, E.; et al. Selective cytotoxicity of a bicyclic Ras inhibitor in cancer cells expressing K-RasG13D. Biochem. Biophys. Res. Commun. 2009, 386, 593–597, doi:10.1016/j.bbrc.2009.06.069.
- Johnson, C.W.; Lin, Y.-J.; Reid, D.; Parker, J.; Pavlopoulos, S.; Dischinger, P.; Graveel, C.; Aguirre, A.J.; Steensma, M.; Haigis, K.M.; et al. Isoform-Specific Destabilization of the Active Site Reveals a Molecular Mechanism of Intrinsic Activation of KRas G13D. Cell Rep. 2019, 28, 1538-1550.e7, doi:10.1016/j.celrep.2019.07.026.
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 2016, 6, 1–15, doi:10.1038/srep21949.
- Rabara, D.; Tran, T.H.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 22122–22131, doi:10.1073/pnas.1908353116.
- Li, S.; Balmain, A.; Counter, C.M. A model for RAS mutation patterns in cancers: finding the sweet spot. Nat. Rev. Cancer 2018, 18, 767–777, doi:10.1038/s41568-018-0076-6.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144.
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9.
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707, doi:10.1016/j.cell.2008.08.021.
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670, doi:10.1016/j.cell.2012.01.058.
- Chiaradonna, F.; Sacco, E.; Manzoni, R.; Giorgio, M.; Vanoni, M.; Alberghina, L. Ras-dependent carbon metabolism and transformation in mouse fibroblasts. Oncogene 2006, 25, doi:10.1038/sj.onc.1209528.
- Warburg, O. On the origin of cancer cells. Science (80-. ). 1956, 123, 309–314, doi:10.1126/science.123.3191.309.
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129, doi:10.1038/s42255-020-0172-2.
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 1–15, doi:10.1038/msb.2011.56.
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105, doi:10.1038/nature12040.
- Deberardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324, doi:10.1038/onc.2009.358.
- De Sanctis, G.; Spinelli, M.; Vanoni, M.; Sacco, E. K-ras activation induces differential sensitivity to sulfur amino acid limitation and deprivation and to oxidative and anti-oxidative stress in mouse fibroblasts. PLoS One 2016, 11, doi:10.1371/journal.pone.0163790.
- Baracca, A.; Chiaradonna, F.; Sgarbi, G.; Solaini, G.; Alberghina, L.; Lenaz, G. Mitochondrial Complex I decrease is responsible for bioenergetic dysfunction in K-ras transformed cells. Biochim. Biophys. Acta - Bioenerg. 2010, 1797, 314–323, doi:10.1016/j.bbabio.2009.11.006.
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 8788–8793, doi:10.1073/pnas.1003428107.
- Stewart, D.R.H.; Clark, G.J. Pumping the brakes on RAS-negative regulators and death effectors of RAS. J. Cell Sci. 2020, 133.
- Gorfe, A.A.; Cho, K.J. Approaches to inhibiting oncogenic K-Ras. Small GTPases 2019, 0, 1–10, doi:10.1080/21541248.2019.1655883.
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017, 168, 878-889.e29, doi:10.1016/j.cell.2017.02.006.
- Ni, D.; Li, X.; He, X.; Zhang, H.; Zhang, J.; Lu, S. Drugging K-RasG12C through covalent inhibitors: Mission possible? Pharmacol. Ther. 2019, 202, 1–17, doi:10.1016/j.pharmthera.2019.06.007.
- Choi, J.A.; Kim, J.Y.; Lee, J.Y.; Kang, C.M.; Kwon, H.J.; Yoo, Y.D.; Kim, T.W.; Lee, Y.S.; Lee, S.J. Induction of cell cycle arrest and apoptosis in human breast cancer cells by quercetin. Int. J. Oncol. 2001, 19, doi:10.3892/ijo.19.4.837.
- Yang, F.Q.; Liu, M.; Li, W.; Che, J.P.; Wang, G.C.; Zheng, J.H. Combination of quercetin and hyperoside inhibits prostate cancer cell growth and metastasis via regulation of microRNA-21. Mol. Med. Rep. 2015, 11, doi:10.3892/mmr.2014.2813.
- Zhang, X.; Guo, Q.; Chen, J.; Chen, Z. Quercetin enhances cisplatin sensitivity of human osteosarcoma cells by modulating microRNA-217-KRAS axis. Mol. Cells 2015, 38–38, doi:10.14348/molcells.2015.0037.
- Ranelletti, F.O.; Maggiano, N.; Serra, F.G.; Ricci, R.; Larocca, L.M.; Lanza, P.; Scambia, G.; Fattorossi, A.; Capelli, A.; Piantelli, M. Quercetin inhibits p21-ras expression in human colon cancer cell lines and in primary colorectal tumors. Int. J. Cancer 2000, 85, doi:10.1002/(SICI)1097-0215(20000201)85:3<438::AID-IJC22>3.0.CO;2-F.
- Garrido, C.M.; Henkels, K.M.; Rehl, K.M.; Liang, H.; Zhou, Y.; Gutterman, J.U.; Cho, K. jin Avicin G is a potent sphingomyelinase inhibitor and blocks oncogenic K- and H-Ras signaling. Sci. Rep. 2020, 10, doi:10.1038/s41598-020-65882-5.
- Pettit, G.R.; Herald, C.L.; Doubek, D.L.; Herald, D.L.; Arnold, E.; Clardy, J. Isolation and Structure of Bryostatin 1. J. Am. Chem. Soc. 1982, 104, doi:10.1021/ja00388a092.
- Kortmansky, J.; Schwartz, G.K. Bryostatin-1: A Novel PKC Inhibitor in Clinical Development. Cancer Invest. 2003, 21.
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.M.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G.; et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 2006, 21, doi:10.1016/j.molcel.2006.01.012.
- Raghuvanshi, R.; Bharate, S.B. Preclinical and Clinical Studies on Bryostatins, A Class of Marine-Derived Protein Kinase C Modulators: A Mini-Review. Curr. Top. Med. Chem. 2020, 20, doi:10.2174/1568026620666200325110444.
- Wang, M.T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell 2015, 163, 1237–1251, doi:10.1016/j.cell.2015.10.041.
- Saha, B.; Nandi, D. Farnesyltransferase inhibitors reduce Ras activation and ameliorate acetaminophen-induced liver injury in mice. Hepatology 2009, 50, doi:10.1002/hep.23180.
- Nagase, T.; Kawata, S.; Tamura, S.; Matsuda, Y.; Inui, Y.; Yamasaki, E.; Ishiguro, H.; Ito, T.; Miyagawa, J.; Mitsui, H.; et al. Manumycin and gliotoxin derivative KT7595 block Ras farnesylation and cell growth but do not disturb lamin farnesylation and localization in human tumour cells. Br. J. Cancer 1997, 76, doi:10.1038/bjc.1997.499.
- Hara, M.; Akasaka, K.; Akinaga, S.; Okabe, M.; Nakano, H.; Gomez, R.; Wood, D.; Uh, M.; Tamanoi, F. Identification of Ras farnesyltransferase inhibitors by microbial screening. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, doi:10.1073/pnas.90.6.2281.
- Lantry, L.E.; Zhang, Z.; Crist, K.A.; Wang, Y.; Hara, M.; Zeeck, A.; Lubet, R.A.; You, M. Chemopreventive efficacy of promising farnesyltransferase inhibitors. In Proceedings of the Experimental Lung Research; 2000; Vol. 26.
- Kouchi, H.; Nakamura, K.; Fushimi, K.; Sakaguchi, M.; Miyazaki, M.; Ohe, T.; Namba, M. Manumycin A, inhibitor of ras farnesyltransferase, inhibits proliferation and migration of rat vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1999, 264, doi:10.1006/bbrc.1999.1546.
- Datta, A.; Kim, H.; Lal, M.; McGee, L.; Johnson, A.; Moustafa, A.A.; Jones, J.C.; Mondal, D.; Ferrer, M.; Abdel-Mageed, A.B. Manumycin A suppresses exosome biogenesis and secretion via targeted inhibition of Ras/Raf/ERK1/2 signaling and hnRNP H1 in castration-resistant prostate cancer cells. Cancer Lett. 2017, 408, doi:10.1016/j.canlet.2017.08.020.
- Chowdhury, R.; Webber, J.P.; Gurney, M.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger mesenchymal stem cell differentiation into pro-angiogenic and pro-invasive myofibroblasts. Oncotarget 2015, 6, doi:10.18632/oncotarget.2711.
- Gelb, M.H.; Tamanoi, F.; Yokoyama, K.; Ghomashchi, F.; Esson, K.; Gould, M.N. The inhibition of protein prenyltransferases by oxygenated metabolites of limonene and perillyl alcohol. Cancer Lett. 1995, 91, doi:10.1016/0304-3835(95)03747-K.
- Chen, X.G.; Shuzo, O.; Li, Y.; Han, R. Inhibition of farnesyl protein transferase, h-ras oncogene expression and p21ras membrane association by natural products in human solid tumor cell lines. J. Asian Nat. Prod. Res. 1998, 1, doi:10.1080/10286029808039842.
- Chaudhary, S.C.; Siddiqui, M.S.; Athar, M.; Alam, M.S. D-Limonene modulates inflammation, oxidative stress and Ras-ERK pathway to inhibit murine skin tumorigenesis. Hum. Exp. Toxicol. 2012, 31, doi:10.1177/0960327111434948.
- Afshordel, S.; Kern, B.; Clasohm, J.; König, H.; Priester, M.; Weissenberger, J.; Kögel, D.; Eckert, G.P. Lovastatin and perillyl alcohol inhibit glioma cell invasion, migration, and proliferation - Impact of Ras-/Rho-prenylation. Pharmacol. Res. 2015, 91, doi:10.1016/j.phrs.2014.11.006.
- Singh, S.B.; Zink, D.L.; Liesch, J.M.; Ball, R.G.; Goetz, M.A.; Bolessa, E.A.; Giacobbe, R.A.; Silverman, K.C.; Bills, G.F.; Pelaez, F.; et al. Preussomerins and Deoxypreussomerins: Novel Inhibitors of Ras Farnesyl-Protein Transferase. J. Org. Chem. 1994, 59, doi:10.1021/jo00100a035.
- Weber, H.A.; Baenziger, N.C.; Gloer, J.B. Structure of Preussomerin A: An Unusual New Antifungal Metabolite from the Coprophilous Fungus Preussia isómera. J. Am. Chem. Soc. 1990, 112, doi:10.1021/ja00174a045.
- Weber, H.A.; Gloer, J.B. The Preussomerins: Novel Antifungal Metabolites from the Coprophilous Fungus Preussia isomera Cain. J. Org. Chem. 1991, 56, doi:10.1021/jo00014a007.
- Polishook, J.D.; Dombrowski, A.W.; Tsou, N.N.; Salituro, G.M.; Curotto, J.E. Preussomerin D from the Endophyte Hormonema Dematioides . Mycologia 1993, 85, doi:10.1080/00275514.1993.12026246.
- Weerapreeyakul, N.; Anorach, R.; Khuansawad, T.; Yenjai, C.; Isaka, M. Synthesis of bioreductive esters from fungal compounds. Chem. Pharm. Bull. 2007, 55, doi:10.1248/cpb.55.930.
- Vigushin, D.M.; Brooke, G.; Willows, D.; Coombes, R.C.; Moody, C.J. Pyrazino[1,2-a]indole-1,4-diones, simple analogues of gliotoxin, as selective inhibitors of geranylgeranyltransferase I. Bioorganic Med. Chem. Lett. 2003, 13, doi:10.1016/j.bmcl.2003.08.022.
- Omura, S.; Van Der Pyl, D.; Inokoshi, J.; Takahashi, Y.; Takeshima, H. Pepticinnamins, new farnesyl-protein transferase inhibitors produced by an actinomycete i. producing strain, fermentation, isolation and biological activity. J. Antibiot. (Tokyo). 1993, 46, doi:10.7164/antibiotics.46.222.
- Ōmura, S.; Tomoda, H. Microbial metabolites affecting lipid biosynthesis. Pure Appl. Chem. 1994, 66, doi:10.1351/pac199466102267.
- Santa Maria, K.C.; Chan, A.N.; O’Neill, E.M.; Li, B. Targeted Rediscovery and Biosynthesis of the Farnesyl-Transferase Inhibitor Pepticinnamin E. ChemBioChem 2019, 20, doi:10.1002/cbic.201900025.
- Thutewohl, M.; Kissau, L.; Popkirova, B.; Karaguni, I.M.; Nowak, T.; Bate, M.; Kuhlmann, J.; Müller, O.; Waldmann, H. Identification of mono- and bisubstrate inhibitors of protein farnesyltransferase and inducers of apoptosis from a pepticinnamin E library. Bioorganic Med. Chem. 2003, 11, doi:10.1016/S0968-0896(03)00160-3.
- Singh, S.B.; Jayasuriya, H.; Silverman, K.C.; Bonfiglio, C.A.; Williamson, J.M.; Lingham, R.B. Efficient syntheses, human and yeast farnesyl-protein transferase inhibitory activities of chaetomellic acids and analogues. Bioorganic Med. Chem. 2000, 8, doi:10.1016/S0968-0896(99)00312-0.
- Gibbs, J.B.; Pompliano, D.L.; Mosser, S.D.; Rands, E.; Lingham, R.B.; Singh, S.B.; Scolnick, E.M.; Kohl, N.E.; Oliff, A. Selective inhibition of farnesyl-protein transferase blocks Ras processing in vivo. J. Biol. Chem. 1993, 268.
- Nogueira, A.; Vala, H.; Vasconcelos-Nóbrega, C.; Faustino-Rocha, A.I.; Pires, C.A.; Colaço, A.; Oliveira, P.A.; Pires, M.J. Long-term treatment with chaethomellic acid A reduces glomerulosclerosis and arteriolosclerosis in a rat model of chronic kidney disease. Biomed. Pharmacother. 2017, 96, doi:10.1016/j.biopha.2017.09.137.
- Oh, H.M.; Choi, S.K.; Lee, J.M.; Lee, S.K.; Kim, H.Y.; Han, D.C.; Kim, H.M.; Son, K.H.; Kwon, B.M. Cyclopentenediones, inhibitors of farnesyl protein transferase and anti-tumor compounds, isolated from the fruit of Lindera erythrocarpa Makino. Bioorganic Med. Chem. 2005, 13, doi:10.1016/j.bmc.2005.06.029.
- Yoon, J.H.; Pham, T.H.; Lee, J.; Lee, J.; Ryu, H.W.; Oh, S.R.; Oh, J.W.; Yoon, D.Y. Methyl linderone suppresses TPA-stimulated IL-8 and MMP-9 expression via the ERK/STAT3 pathway in MCF-7 breast cancer cells. J. Microbiol. Biotechnol. 2020, 30, doi:10.4014/jmb.1911.11068.
- Costa, S.M.O.; Lemos, T.L.G.; Pessoa, O.D.L.; Pessoa, C.; Montenegro, R.C.; Braz-Filho, R. Chemical constituents from Lippia sidoides and cytotoxic activity. J. Nat. Prod. 2001, 64, doi:10.1021/np0005917.
- Cadelis, M.M.; Bourguet-Kondracki, M.L.; Dubois, J.; Valentin, A.; Barker, D.; Copp, B.R. Discovery and preliminary structure-activity relationship studies on tecomaquinone i and tectol as novel farnesyltransferase and plasmodial inhibitors. Bioorganic Med. Chem. 2016, 24, doi:10.1016/j.bmc.2016.05.024.
- Angamuthu, V.; Shanmugavadivu, M.; Nagarajan, G.; Velmurugan, B.K. Pharmalogical activities of antroquinonol- Mini review. Chem. Biol. Interact. 2019, 297.
- Ho, C.L.; Wang, J.L.; Lee, C.C.; Cheng, H.Y.; Wen, W.C.; Cheng, H.H.Y.; Chen, M.C.M. Antroquinonol blocks Ras and Rho signaling via the inhibition of protein isoprenyltransferase activity in cancer cells. Biomed. Pharmacother. 2014, 68, doi:10.1016/j.biopha.2014.09.008.
- Liu, L.J.; Wang, W.; Huang, S.Y.; Hong, Y.; Li, G.; Lin, S.; Tian, J.; Cai, Z.; Wang, H.M.D.; Ma, D.L.; et al. Inhibition of the Ras/Raf interaction and repression of renal cancer xenografts in vivo by an enantiomeric iridium(III) metal-based compound. Chem. Sci. 2017, 8, 4756–4763, doi:10.1039/c7sc00311k.
- Kirn, M.J.; Bok, S.H.; Kwon, B.M.; Shin, J.; Seo, Y. Arteminolide, an inhibitor of farnesyl transferase from Artemisia sylvatica. J. Org. Chem. 1998, 63, doi:10.1021/jo980919p.
- Lee, S.H.; Seo, Y.; Kim, H.K.; Kang, H.M.; Kim, J.H.; Son, K.H.; Lee, H.; Kwon, B.M.; Shin, J.; Seo, J.M. Arteminolides B, C, and D, new inhibitors of farnesyl protein transferase from Artemisia argyi. J. Org. Chem. 2002, 67, doi:10.1021/jo020299z.
- Wen, J.; Shi, H.; Xu, Z.; Chang, H.; Jia, C.; Zan, K.; Jiang, Y.; Tu, P. Dimeric guaianolides and sesquiterpenoids from Artemisia anomala. J. Nat. Prod. 2010, 73, doi:10.1021/np900462u.
- Lin, H.C.; Lin, M.H.; Liao, J.H.; Wu, T.H.; Lee, T.H.; Mi, F.L.; Wu, C.H.; Chen, K.C.; Cheng, C.H.; Lin, C.W. Antroquinonol, a ubiquinone derivative from the mushroom antrodia camphorata, inhibits colon cancer stem cell-like properties: Insights into the molecular mechanism & inhibitory targets. J. Agric. Food Chem. 2017, 65, doi:10.1021/acs.jafc.6b04101.
- Lee, S.H.; Lee, M.Y.; Kang, H.M.; Han, D.C.; Son, K.H.; Yang, D.C.; Sung, N. Do; Lee, C.W.; Kim, H.M.; Kwon, B.M. Anti-tumor activity of the farnesyl-protein transferase inhibitors arteminolides, isolated from Artemisa. Bioorganic Med. Chem. 2003, 11, doi:10.1016/j.bmc.2003.08.008.
- Gbelcová, H.; Rimpelová, S.; Knejzlík, Z.; Šáchová, J.; Kolář, M.; Strnad, H.; Repiská, V.; D’Acunto, W.C.; Ruml, T.; Vítek, L. Isoprenoids responsible for protein prenylation modulate the biological effects of statins on pancreatic cancer cells. Lipids Health Dis. 2017, 16, doi:10.1186/s12944-017-0641-0.
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13.
- Pratheeshkumar, P.; Sreekala, C.; Zhang, Z.; Budhraja, A.; Ding, S.; Son, Y.; Wang, X.; Hitron, A.; Hyun-jung, K.; Wang, L.; et al. Cancer Prevention with Promising Natural Products: Mechanisms of Action and Molecular Targets. 2015, 16, 461–470, doi:10.1016/j.chembiol.2009.02.014.A.
- Tewari, D.; Patni, P.; Bishayee, A.; Sah, A.N.; Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 2019.
- Kumar, S.; Agnihotri, N. Piperlongumine, a piper alkaloid targets Ras/PI3K/Akt/mTOR signaling axis to inhibit tumor cell growth and proliferation in DMH/DSS induced experimental colon cancer. Biomed. Pharmacother. 2019, 109, doi:10.1016/j.biopha.2018.10.182.
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18.
- Zhang, X.; Chen, L.X.; Ouyang, L.; Cheng, Y.; Liu, B. Plant natural compounds: Targeting pathways of autophagy as anti-cancer therapeutic agents. Cell Prolif. 2012, 45, 466–476, doi:10.1111/j.1365-2184.2012.00833.x.
- Alqathama, A.; Prieto, J.M. Natural products with therapeutic potential in melanoma metastasis. Nat. Prod. Rep. 2015, 32, 1170–1182, doi:10.1039/c4np00130c.
- Sacco, E.; Abraham, S.J.; Palmioli, A.; Damore, G.; Bargna, A.; Mazzoleni, E.; Gaponenko, V.; Vanoni, M.; Peri, F. Binding properties and biological characterization of new sugar-derived Ras ligands. Medchemcomm 2011, 2, 396–401, doi:10.1039/c0md00264j.
- Palmioli, A.; Sacco, E.; Abraham, S.; Thomas, C.J.; Domizio, A. Di; Gioia, L. De; Gaponenko, V.; Vanoni, M.; Peri, F. First experimental identification of Ras-inhibitor binding interface using a water-soluble Ras ligand. Bioorganic Med. Chem. Lett. 2009, 19, 4217–4222, doi:10.1016/j.bmcl.2009.05.107.
- Palmioli, A.; Ciaramelli, C.; Tisi, R.; Spinelli, M.; De Sanctis, G.; Sacco, E.; Airoldi, C. Natural Compounds in Cancer Prevention: Effects of Coffee Extracts and Their Main Polyphenolic Component, 5-O-Caffeoylquinic Acid, on Oncogenic Ras Proteins. Chem. - An Asian J. 2017, 12, doi:10.1002/asia.201700844.
- Sturm, S.; Gil, R.R.; Chai, H.B.; Ngassapa, O.D.; Santisuk, T.; Reutrakul, V.; Howe, A.; Moss, M.; Besterman, J.M.; Yang, S.L.; et al. Lupane derivatives from Lophopetalum wallichii with farnesyl protein transferase inhibitory activity. J. Nat. Prod. 1996, 59, doi:10.1021/np960370u.
- Ganaie, A.A.; Siddique, H.R.; Sheikh, I.A.; Parray, A.; Wang, L.; Panyam, J.; Villalta, P.W.; Deng, Y.; Konety, B.R.; Saleem, M. A novel terpenoid class for prevention and treatment of KRAS-driven cancers: Comprehensive analysis using in situ, in vitro, and in vivo model systems. Mol. Carcinog. 2020, 59, doi:10.1002/mc.23200.
- Kim, C.K.; Wang, D.; Bokesch, H.R.; Fuller, R.W.; Smith, E.; Henrich, C.J.; Durrant, D.E.; Morrison, D.K.; Bewley, C.A.; Gustafson, K.R. Swinhopeptolides A and B: Cyclic Depsipeptides from the Sponge Theonella swinhoei That Inhibit Ras/Raf Interaction. J. Nat. Prod. 2020, 83, doi:10.1021/acs.jnatprod.0c00136.
- Thornburg, C.C.; Britt, J.R.; Evans, J.R.; Akee, R.K.; Whitt, J.A.; Trinh, S.K.; Harris, M.J.; Thompson, J.R.; Ewing, T.L.; Shipley, S.M.; et al. NCI Program for Natural Product Discovery: A Publicly-Accessible Library of Natural Product Fractions for High-Throughput Screening. ACS Chem. Biol. 2018, 13, doi:10.1021/acschembio.8b00389.
- Gu, J.; Gui, Y.; Chen, L.; Yuan, G.; Lu, H.Z.; Xu, X. Use of Natural Products as Chemical Library for Drug Discovery and Network Pharmacology. PLoS One 2013, 8, doi:10.1371/journal.pone.0062839.