Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Wendy Huang and Version 1 by Haleema Ahmad.
The PROteolysis TArgeting Chimeras (PROTAC), a technology for targeted protein degradation, relies on using heterobifunctional molecules to recruit intracellular protein degradation machinery to the intracellular target protein of interest. This chemically-induced proximity between protein degradation machinery and the target POI results in polyubiquitylation and proteasomal degradation of the target protein. Despite the field of PROTAC technology being relatively new, PROTACs have found wide applications not just as a technical tool but also as a therapeutic approach for infectious and non-infectious diseases, including cancer and neurodegenerative diseases.
- PROteolysis TArgeting Chimeras
- viruses
- antiviral
- vaccine
1. Introduction
Viruses caused massive deaths during the 20th century; for instance, smallpox killed up to 400 million people [57][1], and influenza caused around 100 million deaths during the great Spanish flu outbreak in 1918–1919 [58][2]. Since it was initially identified in 1981, the human immunodeficiency virus (HIV) epidemic has been responsible for 35 million fatalities [59,60][3][4]. As of 25 January 2022, confirmed cases of COVID-19 had reached 352 million, with 5.60 million deaths (https://covid19.who.int, accessed on 28 November 2022). This pandemic was caused by the SARS-CoV-2 and is referred to as “among the deadliest pandemics of the past century” [61,62][5][6]. Viral infections have become a serious concern to public health and safety. Currently, the prevention and treatment of human viral infections mainly rely on a combination of drugs and vaccines [63,64][7][8]. However, growing drug-resistant strains pose a challenge for currently available antiviral therapeutic strategies, while vaccinations frequently fail to protect against altered or novel viruses [65][9]. Therefore, it is crucial to find cutting-edge antiviral therapeutic strategies based on either novel drug targets or innovative targeting or vaccination strategies. As discussed above, PROTAC technology has been widely investigated for the targeted protein degradation of POI in the context of cancer. But, of late, many advances have been made in understanding the role of PROTACs as antiviral therapeutics. Various PROTAC-based antiviral therapeutic strategies with improved resistance profiles have been recently explored. By putting these novel approaches into practice, it may be possible to develop powerful antiviral therapeutics capable of combating the current and future risks posed by emerging and re-emerging viral pandemics.
2. PROTAC Virus, a Novel Vaccine Strategy
A vaccine is a biological preparation of attenuated or killed disease-causing microorganisms that has the potential to elicit an immune response. Vaccines may also be formulated using toxins released by such microorganisms or their surface receptors that mimic their identity in the host body. Vaccines induce an immune response upon detecting a foreign entity unfamiliar to the body and hence help develop humoral immunity. They are of various types: attenuated, inactivated, toxoid, subunit, conjugate, heterotypic, and genetic [66][10]. Conventionally, live attenuated vaccines have been employed to prevent influenza infections. But they are limited by suboptimal immunogenicity, safety concerns, and cumbersome manufacturing processes [67][11].
Recently, Si et al. described a novel PROTAC-based approach to generate engineered attenuated influenza virus strains for vaccination purposes. They employ PROTAC technology for targeted protein degradation of selected viral proteins of the influenza virus by the host ubiquitin-proteasome system, dramatically attenuating viral replication. Although PROTAC viruses were defective in their replication, they could elicit robust immune responses. To generate attenuated influenza PROTAC viruses, they engineered the genome of influenza A by linking the proteasome targeting domain (PTD) to the matrix gene segments. The PTD, a heptapeptide with a sequence ALAPYIP, is recognized by VHL E3 ubiquitin ligase. The tagging of viral proteins by PTD results in their polyubiquitylation by VHL E3 ligase, followed by proteolysis by the host ubiquitin-proteasome system. As PTD peptide is linked to the viral protein through a tobacco etch virus (TEV) cleavage site linker peptide (ENLYFQG), it can be conditionally cleaved in cell lines that stably express TEV protease, sparing the proteolysis of the PTD-tagged target proteins. Therefore, influenza virus replication was not defective in cell lines stably expressing TEV, producing attenuated virus particles for vaccine manufacture.
Si. et al. tested eight different influenza A proteins (M1, PB2, PB1, PA, NP, M2, NEP, and NS1) for PTD-tagging dependent proteolysis and attenuation of replication in conventional Madin-Darby canine kidney 2 (MDCK2) cells and PTD-cleavage efficiency and protection from proteolysis in TEV expressing MDCK2 cells (MDCK-TEVp). Although PTD-tagging led to efficient proteolysis of each of the eight proteins in MDCK2 cells, the PTD-cleavage efficiency and, thus, the protection from proteolysis in MDCK-TEVp was highly variable. This was largely attributed to the accessibility of the TEV-cleavage site of the PTD-tagged proteins. Among the eight proteins, PTD-tagging of M1 (M1-PTD) showed efficient proteolysis of M1 protein in MDCK2 cells, resulting in >20,000-fold decrement in replication competence compared to wild-type virus. On the contrary, the M1-PTD virus efficiently replicated in TEV expressing MDCK-TEVp cell line (Figure 1). Also, the M1-PTD virus was not only highly attenuated in BALB/c mice and ferrets but was also genetically stable. The group also tested the ability of M1-PTD to induce an immune response in mice and ferrets and detected titters for HI (Hemagglutinin Inhibition), NT (Neutralization), HA (hemagglutinin), and internally conserved nucleoprotein antibodies significantly higher than those immunized with inactivated influenza vaccine (IIV) or by cold-adapted influenza vaccine (CAIV). A robust and adept T-cell immune response was also detected due to the enhanced presentation of degraded viral peptide antigens by MHC molecules.
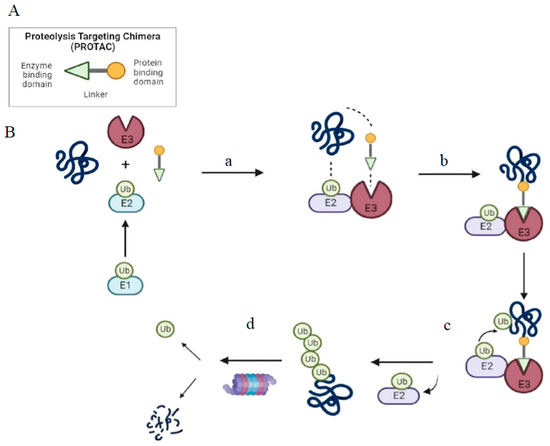
Figure 1.
Structure and mechanism of PROTAC-based degradation. (
A
) General structure of a PROTAC: The E3 ligase targeting “anchor” (green) is connected to the specific POI targeting warhead (yellow) via a variable linker; (
B
) Mechanism of PROTAC-mediated target degradation (a) Ub transfer from E1 to E2, which is followed by complex formation with an E3 ligase; (b) the PROTAC binds to both the E3 ligase and POI to form a TC. This brings the E2 ligase into proximity to the POI; (c) this leads to the transfer of multiple Ub units to surface-exposed lysine residues; (d) the resulting polyubiquitin chain is recognized by the proteasome, leading to the proteolytic degradation of the POI. Ub, Ubiquitin; POI, Protein of interest; TC, Ternary complex.
PROTAC viruses hold the potential as an ideal vaccine candidate. An ideal vaccine is capable of attaining a level of sufficient attenuation in the host for safety while also retaining robust immunogenicity in cell lines [68,69][12][13]. In contrast to the conventional methodologies of vaccine production, PROTAC exploits the degraded viral peptides generated from the proteasomal degradation pathway to trigger an efficient immune response [70][14]. PROTAC technology is more efficient than other methodologies of attenuation due to improved safety and a cumulative loss in efficacy or productivity. Another prominent challenge to the conventional attenuation-based approaches is the immune escape due to rapid viral evolution. Hence, PROTAC technology has emerged as a prominent option for generating safer and more effective vaccines.
3. Proteases-Targeting PROTAC
The viruses encode one or more proteases as a typical technique to support replication with a compacted genome. To produce mature viral proteins, the viral genome encodes a polyprotein with an integrated viral protease that cleaves the polyprotein at several particular places. As a result of their necessity for reproduction, viral proteases are excellent therapeutic targets. The viral life cycle and replication depend on the viral proteases. Proteases from various viral families differ from one another in terms of structure, catalytic mechanism, and preferred substrate. Viral proteases have a specific substrate preference that can be used when designing inhibitors to create strong and selective drug-like compounds. Extensive studies on the role of viral proteases hint towards their important role in the replication phase of viruses. Hence, viral proteases are a common target for inhibiting viral replication in antiviral strategies. Various conventional small molecule inhibitors against viral proteases have been developed over the last few decades. In order to overcome the drug resistance as well as improve the efficacy of the anti-protease small molecule inhibitors, some researchers applied PROTAC-based target protein degradation of viral proteases.
A PROTAC compound against a non-structural 3/4A (NS3/4A) serine protease of the hepatitis C virus (HCV) was described [71][15]. Telaprevir, a reversible covalent inhibitor that binds to the active site of the HCV NS3/4A serine protease, was used to develop a PROTAC degrader for this protease (Figure 2). Telaprevir’s crystal structure with the viral protease revealed that its pyrazine ring is solvent exposed, allowing it to be derivatized with various linkers conjugated to ligands of CRBN, the substrate receptor for the CUL4-RBX1-DDB1-CRBN E3 ubiquitin ligase. The resulting telaprevir-based bivalent degrader, DGY-08-097, caused both inhibition and selective degradation of the HCV NS3/4A protease in cellular infection models. Additional PROTAC molecules based on boceprevir, a synthetic tripeptide that selectively inhibits NS3/4A protease, can be developed as a potential antiviral against HCV.
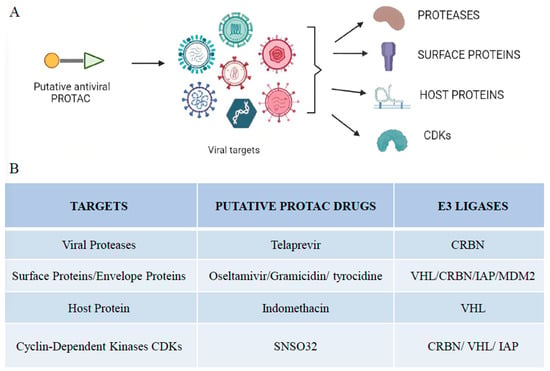
Figure 2.
Viral targets of different PROTAC-based approaches. (
A
) Illustration of putative antiviral PROTACs targeting different viral targets, including proteases, surface proteins, host proteins, and cyclin-dependent kinases. (
B
) Summary of viral targets and their putative antiviral drugs and E3 ligases that make up the PROTAC molecule.
4. Surface Receptor-Targeting PROTACs
The viral envelope is a lipid bilayer that encapsulates the capsid. It is embedded with many glycoproteins on its surface, enabling the virus to attach to the host cell’s receptors. On the other hand, many coronaviruses (CoVs) possess unique envelope and membrane proteins essential for the sustenance of viral replication, apart from these glycoproteins present in the envelope. The viral surface proteins, like hemagglutinin and neuraminidase, are emerging as the new target for antiviral therapies due to their stark difference from the receptors on the host’s cell surface. Hemagglutinin is a lectin involved in the virus’s attachment to its receptor on the cell surface by recognizing terminal sialic acid residues present on them [75][16]. Neuraminidase helps move the virus from one sialic acid residue to another by cleaving the bond between hemagglutinin present on the viral envelope and sialic acid residues on the host cell membrane [76][17]. The cycle continues, and the virus traverses the host’s cell membrane until it recognizes and attaches to the proper cell receptor for its entry into the host cell. Similarly, the activity of neuraminidase also makes it possible for freshly created virions to leave the surface of the host cell. Any damage to the viral envelope may irreversibly damage it due to the generation of reduced oxygen species because the viruses lack the machinery to repair them [77,78][18][19].
Oseltamivir, a neuraminidase inhibitor, prevents the exit of the newly synthesized viral particles from the host cell. Neuraminidase is involved in cleaving the bond of hemagglutinin with sialic acid residues that attach the newly synthesized virions to the host cell’s surface. It has been widely used in the treatment of infections caused by influenza A and influenza B viruses. Oseltamivir-based PROTACs against the influenza virus were developed by connecting E3 ligase ligands on the N-terminal and carboxyl-terminal of Oseltamivir via diversified linkers (Figure 2). Hydrophobic interactions and hydrogen bonds of the synthesized PROTAC molecules with neuraminidase and VHL ligase were involved in the formation of a stable ternary complex. The N-substituted oseltamivir-based PROTACs were more effective than their carboxylate counterparts [79][20]. The mechanism of action of oseltamivir-based PROTACs involves the targeting and degradation of neuraminidase via the ubiquitin-proteasome pathway. The oseltamivir-based PROTACs tend to work dually. Firstly, these PROTACs have oseltamivir connected to one end, which has a strong affinity for neuraminidase and inhibits its action. Secondly, these PROTACs employ the E3 ligases to degrade the neuraminidase enzyme. Inhibition of the neuraminidase activity and its subsequently degradation ensures that the virions synthesized in the host cell do not leave and remain attached to the host cell. These PROTACs were effective against oseltamivir-resistant strains on further evaluation [79][20].
Apart from targeting the most commonly present glycoproteins in the envelope of viruses, certain other envelope and membrane proteins can also be targeted for degradation that can subsequently hinder viral productivity, as in the case of coronaviruses. Coronaviruses (CoVs) are enveloped viruses containing a positive, single-stranded RNA genome packaged within a capsid. The capsid consists of the nucleocapsid protein N, which is further surrounded by a membrane that contains three proteins: the membrane protein (M) and the envelope protein (E), which are involved in the virus budding process, and the spike glycoprotein (S), which is a key player in binding the host receptor and mediating membrane fusion and virus entry into host cells [80,81][21][22]. The envelope protein E within the SARS-CoV-2 can be a potential target protein of interest. Many factors make this protein feasible as a target protein. The protein E is the least abundant among the other proteins and is the only protein that is not glycosylated [82][23]. As there is a lack of glycosylation, the protein can be easily accessible to small molecules to engage with it. Since its absence or inactivation can directly alter viral assembly, membrane permeabilizing activity, and other parameters, targeting envelope protein E affects virulence. Additionally, viruses frequently associate with receptor proteins on cell surfaces to enter human cells; for instance, the SARS-CoV-2 interacts with the angiotensin-converting enzyme 2 (ACE2) receptor [83][24].
PROTAC technology may be an effective method to degrade these proteins and subsequently stop the virus for those targets that are hard to discover direct inhibitors for or non-druggable targets, such as Nsp1, Nsp3b, Nsp3c, E-channel, etc. The drugs having the highest potential binding affinities for the SARS-CoV-2 proteins are gramicidin S and tyrocidine A. Through ACE-2 receptors, S-glycoprotein is crucial for coronavirus attachment to the host cell surface. The association between S-glycoprotein and ACE-2 was broken in binding experiments with Dactinomycin and Gramicidin S to S-glycoprotein, breaking the link between viral S-glycoprotein and the host’s ACE-2 receptor [84][25]. These molecules could be used to design potential PROTAC molecules to target surface proteins. For the S protein, only one compound, natural hesperidin, was found to target the binding between the S receptor binding domain (RBD) and human ACE2 [85][26]. Any small molecule bound to S may interfere with S’s re-folding, inhibiting the viral infection process.
Furthermore, small molecules targeting any part of the S protein may be a good starting point for designing PROTAC-based therapy. Aside from the S protein, the E protein (E-channel) performs crucial biological tasks for the coronavirus’s structural integrity and host pathogenicity. For N proteins in host cells to bind with coronavirus RNA effectively, they require the NRBD and CRBD of the coronavirus N protein. Therefore, the NRBD and CRBD domains of the E protein or the N protein can be exploited as targets for developing antiviral medications. The PROTAC against protein E promotes proteasomal degradation. The antiviral PROTAC also has the additional advantage of providing an antiviral immune response. The innate immune response against the viral infection will promote its clearance through its presentation by MHC-I, its proteasomal degradation and development of T-cell antibodies. An increased presentation of viral proteins through MHC-I may lead to increased T-cell activity against the viral protein.
5. Host Protein-Targeting PROTAC
After the infection of the host cell with the virus, the viruses hijack the host cell’s machinery to synthesize viral proteins and nucleic acids for their rapid multiplication and assembly. Several enzymes are necessary for viral replication and productivity. These include polymerases, endonucleases, and ligases involved in various nucleic acid transactions. A new shift in the development of antiviral therapeutics has been seen in seeking host proteins as targets. It will enable tackling the resurgence of drug resistance in viruses. Furthermore, since viral pathogens require host machinery to replicate their genetic material, targeting host proteins successfully will help bypass the hurdle of frequently tweaking antivirals frequently to effectively target the rapidly mutating viral genome.
Drug repurposing was the most sought-after strategy to tackle the COVID-19 pandemic by identifying existing drugs as effective antivirals. One such drug was indomethacin (INM), which was discovered as a potential antiviral drug candidate using a network-based deep-learning methodology and omics-based analysis. INM is a non-steroid anti-inflammatory drug with analgesic and antipyretic properties and is known to inhibit PGES-2 (prostaglandin E synthase type-2), an enzyme known to be involved in eicosanoid biosynthesis [86][27]. It was shown that INM inhibits the SARS-CoV-2 replication cycle by activating the protein kinase R pathway, inhibiting protein synthesis in virus-infected cells. INM inhibits the host enzyme PGES-2, which interacts with non-structural protein (NSP7), an essential component of the viral primase complex along with NSP8. The complex is a part of the viral RNA polymerase machinery and is conserved in many variants of CoVs [87,88][28][29]. Hence, targeted protein degradation of PGES-2 by PROTACs may act as a universal anti-CoV drug.
INM-based PROTACs were synthesized by the conjugation of INM with the VHL E3 ligase ligand through an aliphatic or polyethylene glycol linker [89][30]. Since INM-based PROTACs target the host protein (PGES-2), their efficacy as antiviral therapeutics is eminent in overcoming drug resistance issues, usually due to the resurgence of variations in the genome of viral proteins. PROTACs based on INM have been developed as an effective antiviral therapy that inhibits not only the enzymatic activity of the proteins but also their scaffolding activity, downregulating the subsequent cascade of reactions that could otherwise revive viral synthesis despite inhibiting the viral proteins required for replication (Figure 3).
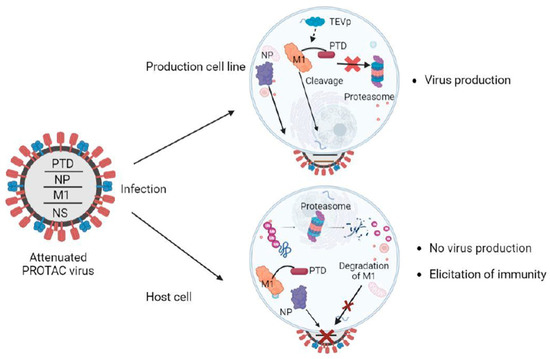
Figure 3.
Overview of the PROTAC-based vaccine design. The ubiquitinated viral protein (M1-PTD) of PROTAC viruses is targeted and degraded by the proteasome in host cells, attenuating their reproduction and causing a deficit in protein synthesis. The influenza nucleoprotein (NP), which is not fused to PTD, is not degraded. In the production cell line, cells are designed to produce the TEV protease (TEVp), which cleaves the PTD, and the PROTAC viruses proliferate well for vaccine production. M1, matrix protein; Ub, ubiquitin; PTD, proteasome-targeting domain.
Similarly, it is well established that cyclin-dependent kinases (CDKs) play an important role in the life cycles of not only DNA and RNA viruses [90][31]. Consistently, the inhibitors of CDK1, CDK2, CDK3, CDK4/6, CDK5, CDK7, and CDK9 show potent antiviral activities against a variety of viruses, including HSV, HIV, Human cytomegalovirus (HCMV), and SARS-CoV-2 [90][31]. These findings prompted Hahn et al. to develop THAL-SNS032, a commercially available CDK9-directed PROTAC (Figure 3). This PROTAC is produced by coupling the E3-recruiting unit thalidomide to the CDK inhibitor SNS032. THAL-SNS032 mostly binds and hence causes degradation of CDK9, but also CDKs 1, 2, and 7 [91][32]. In addition, THAL-SNS032 also targets the HCMV-encoded ortholog of CDKs, pUL97, which play important roles in viral replication. HCMV is a significant opportunistic human pathogen widespread worldwide, with seroprevalence ranging from 40% to 95% in the adult population. It was discovered that THAL-SNS032 was sensitive to the HCMV virus, whereas other viruses exhibited insensitive behavior.
Even though a vast majority of approved antiviral drugs target viral proteins, they tend to pose the risk of the emergence of drug-resistant viral strains, limiting the scope of developing efficient pan-viral therapies. This is because some antiviral therapies may tend to work against a particular strain but will not work against another strain with the same target sites due to the introduction of mutations. To counter these, host-directed therapies are currently being explored. Viruses rely heavily on the host machinery to ensure their replication, and targeting these key host targets will assist in hindering the replication cycle of viruses. Additionally, these host targets can be utilized to develop broad-spectrum antiviral medicines because they have a very limited mutation potential in contrast to rapidly evolving viral genomes. Introducing PROTAC technology in the targeted degradation of host proteins will significantly enhance the specificity of the antiviral strategies. Hence, many new host targets can be explored for PROTACs, including the host receptor, CCR5 (chemokine receptor type 5) in the case of HIV type 1, and tight junction proteins like Claudin1 in the case of HCV.
6. Miscellaneous
The Hepatitis B virus (HBV) is known to infect more than a third of the world’s population, and its infection is a major risk factor for the development of hepatocellular carcinoma. The viral infection and productivity are maintained by the X-protein of the virus, which is involved in the transactivation and dysregulation of multiple cancer-associated genes, DNA repair mechanisms, and cell apoptosis [92,93][33][34]. A novel PROTAC has been designed that is capable of simultaneously degrading and inhibiting the function of protein X [94][35]. The N-terminal oligomerization domain was fused to the C-terminal instability domain of the protein X to construct the novel PROTAC. Additionally, it was made cell-permeable by adding a polyarginine cell-penetrating peptide. The X-protein would bind to the oligomerization domain, and the instability domain of the X-protein would tag the protein for proteasomal degradation. All in all, cell-penetrating PROTACs based on the oligomerization and instability domains of the X-protein can antagonize the X-protein and play an important role in causing its elimination. These novel PROTACs can be employed not only as therapeutics in the treatment of Hepatitis B virus infection but also in the prevention of hepatocellular carcinoma [94][35].
Human immunodeficiency virus (HIV) is a retrovirus that causes acquired immunodeficiency syndrome (AIDS) [95][36]. It is also known as human T-cell leukemia (lymphotropic) virus type III (HTLV-III). The majority of HIV viral proteins are made up of accessory proteins, virus-specific enzymes, and structural proteins. All fully functioning viruses have the same well-studied viral host protein interactions, essential for their activity. There are three HIV-related enzymes: reverse transcriptase, protease, and integrase. The multidomain enzyme HIV-1 integrase is necessary to integrate viral DNA into the host genome. Three domains make up the enzyme. The His2Cys2 motif in the N-terminal domain chelates zinc, the catalytic DDE motif in the core domain is necessary for the enzyme’s enzymatic activity, and the SH3-like fold in the C-terminal domain unspecifically binds DNA [96][37]. By causing the breakdown of viral proteins, the host ubiquitin-proteasome pathway (UPP) performs crucial functions in host defense against viruses. HIV-1 Human Interaction Database (available at https://www.ncbi.nlm.nih.gov/genome/viruses/retroviruses/hiv-1/interactions/, accessed on 30 November 2022) is a comprehensive database of HIV-1 human protein interactions that enables the identification of regions involved in virus-host protein interactions that can be used for designing chimeric E3 ligases. Substrates for ubiquitin-proteasome degradation have been successfully established. In the study by Zhang et al., they attempted to design and construct artificial chimeric ubiquitin ligases (E3s) based on known human E3s in order to target HIV-1 integrase for ubiquitin-proteasome pathway-mediated degradation manually. It was designed in a way that a functional chimeric E3 that can target HIV-1 integrase, the substrate-binding domains of various natural E3s were replaced with the HIV-1 integrase binding domain (IBD) of human LEDGF/P75 protein or the enzymatic domains of E3s were linked with the IBD directly and succeeded in creating chimeric E3 146LIS that was capable of targeting HIV-1 NL4-3 integrase for Lys48-specific polyubiquitination and degradation by 26S proteasome [97][38]. The table below categorizes the numerous viral targets that have been targeted for degradation using PROTAC-based antiviral methods (Table 1).
Table 1.
Summary of PROTACs as an antiviral strategy.
| Target | Examples | E3 Ligase | Mechanism of Action | Ref. |
|---|---|---|---|---|
| Viral proteases | ||||
| Telaprevir | CRBN | Telaprevir acts as a protein ligand in the ternary complex and may reversibly bind to and inhibit the viral proteases. | [71][15] | |
| Surface proteins | ||||
|
Oseltamivir | VHL/CRBN | Oseltamivir binds to the neuraminidase enzyme in the viral envelope and inhibits it. The PROTAC molecule employs the cellular ligases for the subsequent degradation of neuraminidase. | [79][20] |
|
||||
|
Thalidomide | VHL | Targets envelop protein E and lead to its degradation via the proteasomal degradation pathway. | |
| Host protein | ||||
|
Indomethacin | Indomethacin binds to the host protein, PGES-2. PGES2 and NSP-7 form the viral primase complex. Targeting PGES-2 for degradation hinders the formation of the viral polymerase. Alternatively, it interacts with NSPs in the viral primase complex and regulates the PKR pathway to inhibit protein synthesis. | [89][30] | |
|
SNS032 | CRBN/IAP/VHL | CDK-directed PROTACs cause the degradation of other essential CDKs that participate in the formation of the nuclear capsid of the virus. | [91][32] |
References
- Meyer, H.; Ehmann, R.; Smith, G.L. Smallpox in the Post-Eradication Era. Viruses 2020, 12, 138.
- Trilla, A.; Trilla, G.; Daer, C. The 1918 “Spanish Flu” in Spain. Clin. Infect. Dis. 2008, 47, 668–673.
- Lu, L.; Su, S.; Yang, H.; Jiang, S. Antivirals with common targets against highly pathogenic viruses. Cell 2021, 184, 1604–1620.
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519–530.
- Morens, D.M.; Fauci, A.S. Emerging Pandemic Diseases: How We Got to COVID-19. Cell 2020, 182, 1077–1092.
- Gao, S.; Huang, T.; Song, L.; Xu, S.; Cheng, Y.; Cherukupalli, S.; Kang, D.; Zhao, T.; Sun, L.; Zhang, J.; et al. Medicinal chemistry strategies towards the development of effective SARS-CoV-2 inhibitors. Acta Pharm. Sin. B 2022, 12, 581–599.
- De Clercq, E. Fifty Years in Search of Selective Antiviral Drugs. J. Med. Chem. 2019, 62, 7322–7339.
- Zhan, P.; Pannecouque, C.; De Clercq, E.; Liu, X. Anti-HIV Drug Discovery and Development: Current Innovations and Future Trends. J. Med. Chem. 2016, 59, 2849–2878.
- Ma, Y.; Frutos-Beltrán, E.; Kang, D.; Pannecouque, C.; De Clercq, E.; Menéndez-Arias, L.; Liu, X.; Zhan, P. Medicinal chemistry strategies for discovering antivirals effective against drug-resistant viruses. Chem. Soc. Rev. 2021, 50, 4514–4540.
- Francis, M.J. Recent Advances in Vaccine Technologies. Vet. Clin. N. Am. Small Anim. Pract. 2018, 48, 231–241.
- Dane, D.S.; Dick, G.W.A.; Briggs, M.; Nelson, R. Vaccination Against Poliomyelitis with Live Virus Vaccines. 5. Neutralizing anti body levels one year after vaccination. Br. Med. J. 1958, 2, 1187–1188.
- Hussain, A.I.; Cordeiro, M.; Sevilla, E.; Liu, J. Comparison of egg and high yielding MDCK cell-derived live attenuated influenza virus for commercial production of trivalent influenza vaccine: In vitro cell susceptibility and influenza virus replication kinetics in permissive and semi-permissive cells. Vaccine 2010, 28, 3848–3855.
- Pica, N.; Palese, P. Toward a Universal Influenza Virus Vaccine: Prospects and Challenges. Annu. Rev. Med. 2013, 64, 189–202.
- Si, L.; Shen, Q.; Li, J.; Chen, L.; Shen, J.; Xiao, X.; Bai, H.; Feng, T.; Ye, A.Y.; Le Li, L.; et al. Generation of a live attenuated influenza A vaccine by proteolysis targeting. Nat. Biotechnol. 2022, 40, 1370–1377.
- De Wispelaere, M.; Du, G.; Donovan, K.A.; Zhang, T.; Eleuteri, N.A.; Yuan, J.C.; Kalabathula, J.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat. Commun. 2019, 10, 3468.
- Suzuki, Y. Sialobiology of Influenza: Molecular Mechanism of Host Range Variation of Influenza Viruses. Biol. Pharm. Bull. 2005, 28, 399–408.
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front. Immunol. 2018, 9, 1581.
- McMahon, H.T.; Gallop, J.L. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 2005, 438, 590–596.
- Wolf, M.; Garcea, R.L.; Grigorieff, N.; Harrison, S.C. Subunit interactions in bovine papillomavirus. Proc. Natl. Acad. Sci. USA 2010, 107, 6298–6303.
- Xu, Z.; Liu, X.; Ma, X.; Zou, W.; Chen, Q.; Chen, F.; Deng, X.; Liang, J.; Dong, C.; Lan, K.; et al. Discovery of oseltamivir-based novel PROTACs as degraders targeting neuraminidase to combat H1N1 influenza virus. Cell Insight 2022, 1, 100030.
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261.
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Jr, R.M.W.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592.
- Pervushin, K.; Tan, E.; Parthasarathy, K.; Lin, X.; Jiang, F.L.; Yu, D.; Vararattanavech, A.; Soong, T.W.; Liu, D.X.; Torres, J. Structure and Inhibition of the SARS Coronavirus Envelope Protein Ion Channel. PLoS Pathog. 2009, 5, e1000511.
- Ge, X.-Y.; Li, J.-L.; Yang, X.-L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538.
- Bansal, P.; Kumar, R.; Singh, J.; Dhanda, S. In silico molecular docking of SARS-CoV-2 surface proteins with microbial non-ribosomal peptides: Identification of potential drugs. J. Proteins Proteom. 2021, 12, 177–184.
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788.
- Yamada, T.; Komoto, J.; Watanabe, K.; Ohmiya, Y.; Takusagawa, F. Crystal Structure and Possible Catalytic Mechanism of Microsomal Prostaglandin E Synthase Type 2 (mPGES-2). J. Mol. Biol. 2005, 348, 1163–1176.
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468.
- Terracciano, R.; Preianò, M.; Fregola, A.; Pelaia, C.; Montalcini, T.; Savino, R. Mapping the SARS-CoV-2–Host Protein–Protein Interactome by Affinity Purification Mass Spectrometry and Proximity-Dependent Biotin Labeling: A Rational and Straightforward Route to Discover Host-Directed Anti-SARS-CoV-2 Therapeutics. Int. J. Mol. Sci. 2021, 22, 532.
- Desantis, J.; Mercorelli, B.; Celegato, M.; Croci, F.; Bazzacco, A.; Baroni, M.; Siragusa, L.; Cruciani, G.; Loregian, A.; Goracci, L. Indomethacin-based PROTACs as pan-coronavirus antiviral agents. Eur. J. Med. Chem. 2021, 226, 113814.
- Gutierrez-Chamorro, L.; Felip, E.; Ezeonwumelu, I.J.; Margelí, M.; Ballana, E. Cyclin-dependent Kinases as Emerging Targets for Developing Novel Antiviral Therapeutics. Trends Microbiol. 2021, 29, 836–848.
- Hahn, F.; Hamilton, S.T.; Wangen, C.; Wild, M.; Kicuntod, J.; Brückner, N.; Follett, J.E.L.; Herrmann, L.; Kheimar, A.; Kaufer, B.B.; et al. Development of a PROTAC-Based Targeting Strategy Provides a Mechanistically Unique Mode of Anti-Cytomegalovirus Activity. Int. J. Mol. Sci. 2021, 22, 12858.
- Murakami, S. Hepatitis B Virus X Protein: Structure, Function and Biology. Intervirology 1999, 42, 81–99.
- Feitelson, M.A.; Duan, L.X. Hepatitis B virus X antigen in the pathogenesis of chronic infections and the development of hepatocellular carcinoma. Am. J. Pathol. 1997, 150, 1141–1157.
- Montrose, K.; Krissansen, G.W. Design of a PROTAC that antagonizes and destroys the cancer-forming X-protein of the hepatitis B virus. Biochem. Biophys. Res. Commun. 2014, 453, 735–740.
- Landesman, S.H.; Ginzburg, H.M.; Weiss, S. The AIDS Epidemic. N. Engl. J. Med. 1985, 312, 521–525.
- Turner, B.G.; Summers, M.F. Structural biology of HIV. J. Mol. Biol. 1999, 285, 1–32.
- Zhang, Z.; Yuan, S.; Xu, S.; Guo, D.; Chen, L.; Hou, W.; Wang, M. Suppression of HIV-1 Integration by Targeting HIV-1 Integrase for Degradation with A Chimeric Ubiquitin Ligase. Virol. Sin. 2021, 36, 424–437.
More