Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Giuseppe Murdaca and Version 2 by Dean Liu.
Although immunotherapy is already a staple of cancer care, many patients may not benefit from these cutting-edge treatments. A crucial field of research now focuses on figuring out how to improve treatment efficacy and assess the resistance mechanisms underlying this uneven response. For a good response, immune-based treatments, in particular immune checkpoint inhibitors, rely on a strong infiltration of T cells into the tumour microenvironment. The severe metabolic environment that immune cells must endure can drastically reduce effector activity. These immune dysregulation-related tumour-mediated perturbations include oxidative stress, which can encourage lipid peroxidation, ER stress, and T regulatory cells dysfunction.
- immune checkpoint
- immune check points inhibitor
- oxidative stress
1. General Considerations on Immunological Checkpoints and Oxidative Stress in Neoplasms
One strategy tumour cells have developed to evade immune surveillance is a changed expression of the immunological checkpoint receptor programmed death receptor 1 (PD-1) and its ligand, PD-L1 [1]. The ligand–receptor interactions that lead to the activation of numerous immunological checkpoints also involve the cytotoxic T lymphocyte-associated protein-4 (CTLA4), PDL2 receptors expressed on immune and tumour cells, and others [2] (Figure 1). When PD-1 on cytotoxic T-cells binds to PD-L1 on the surface of cancer cells, this inhibits T lymphocyte stimulation and immune escape. Probably, the primary method of immunological resistance to cancer is the activation of specific immune checkpoint mechanisms.
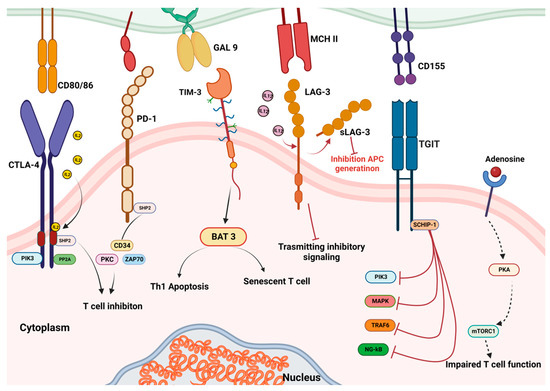
Figure 1. Immune checkpoint structure and interactions.
Immunotherapy reduces immunological tolerance by preventing the communication between tumour cells and the immune system. Hence, it is anticipated that blocking immunological checkpoints will be a new cancer therapeutic method [1]. In fact, to prevent the contact and hence restore antitumour immunity, monoclonal antibodies (mAbs) targeting PD-1 or PDL1 have been produced. Over the past few years, a number of mAbs have been registered in various tumour pathologies, and a number of further anti-PD-(L)1 mAbs are presently undergoing clinical progress. The similar goal of blocking the checkpoint and activating T cell-based immunotreatment has also been achieved via the development of peptides and small compounds that target PD-L1 [3]. Increasing evidence demonstrates that immunotherapy techniques are highly effective at eliminating tumours, preventing their reappearance, and have potential for future application [4].
Immune checkpoint inhibitor (ICI) medications have had significant success; however, most patients who receive ICI monotherapy do not experience sufficient long-term anticancer effects [5]. The so-called “cold tumour” caused by defects in antigen presentation to T cells, absence of T cell activation, lack or minority of activated T cell infiltration in tumour tissues, and abundance of immune suppressor cells such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) is one of the main indicators of a poor response to ICI therapy [6][7][8][9][6,7,8,9].
As a result, recent research on immunogenic cell death (ICD) has been conducted extensively to study the field of cancer immunotherapy [10][11][10,11] and ways of reducing oxidative stress (OS), which led to an improved immune response against the tumour [12]. In fact, OS is one of the most representative biological situations that cancer cells are typically exposed to [13][14][13,14]. The raised intracellular amounts of reactive oxygen species (ROS) are a feature of the cancer milieu, which is characterized by increased OS [15]. ROS are extremely reactive oxygen compounds that include hydrogen peroxide, superoxide, peroxides, and hydroxyl radicals. The activation of specific oncogenes, hypoxia, and external stimuli such as chemotherapy and radiotherapy can all be linked to dysregulated ROS in tumours cells.
Owed to abnormalities in DNA repair, protein degradation, and lipid peroxidation, excessive ROS production might be fatal to cancer cells [16][17][18][16,17,18]. Consequently, OS has a significant function in the emergence and growth of cancers as well as the management of neoplastic illnesses [19]. The intracellular biological redox steady state is disrupted when cells are continuously exposed to environmental stress, such as UV radiation, metabolic stress, and anti-cancer medications. Excessive ROS are then produced, which affects immune dysfunction, signal transduction, cell growth, and cell death [20]. However, as mentioned above, ROS may also cause DNA base modifications or sequence rearrangements, DNA damage-derived miscoding lesions, and oncogene activation, all of which work in concert to promote the growth and spread of tumours [21] (Figure 2). In actuality, OS has a role in a number of malignancies, including those related to the brain, breast, pancreatic adenocarcinoma, lung cancer, and others [22][23][24][25][26][27][28][22,23,24,25,26,27,28].
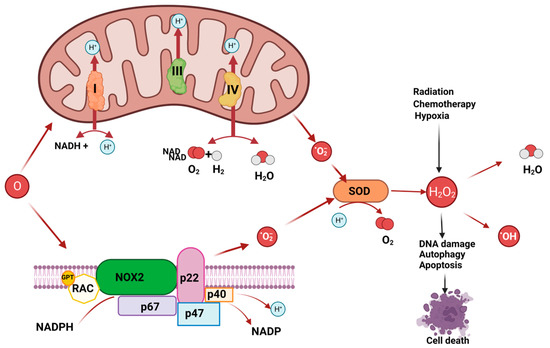
Figure 2. ROS production may derive by mitochondrial respiratory chain and NADPH oxidase or may derive from hypoxia, or chemotherapy. ROS may induce DNA base modifications, DNA damage and cell death.
2. Oxidative Stress and Immune System
Both immune effectors and tumour cells are influenced in a tumour microenvironment (TME) when ROS concentrations are maintained at elevated levels. According to certain studies, immune cells’ ability to act as antioxidants plays a role in their ability to combat cancer [29][30][29,30]. Immune suppression takes place in the TME when the ROS level rises to prevent immune cells from destroying tumours [31]. Furthermore, knowledge of the effects of ROS on dendritic cells (DC), macrophages, natural killer (NK) cells, T cells, and B cells has increased [32][33][34][32,33,34]. Because it affects both tumour cells and TME, oxidative stress has been shown to either promote or inhibit the growth or spread of tumours [35].
Moreover, ROS have a substantial impact on how PD-1 and PD-L1 are expressed, although it is not always clear how ROS and PD-(L)1 interact. Nonetheless, a number of studies have indicated that ROS regulate PD-L1 expression [36]. Particularly important, a considerable increase in the expression of PD-L1 was observed when cells were exposed to a number of ROS inducers, including buthionine sulphoximine and the anticancer drug paclitaxel. [37]. Many natural items, well-known medications, and experimental chemicals were found to modulate the expression of PD-L1 and ROS.
An important component of redox control and antioxidant defence is the thioredoxin (Trx) system. It is made up of the crucial anticancer targets Trx and thioredoxin reductase (TrxR). Anticancer drugs frequently target the Trx/TrxR system. The thi-oredoxin reductase 1 (TrxR1) enzyme, for instance, is a vital intracellular redox sensor and antioxidant enzyme that is frequently overexpressed in a range of cancer types. It is inhibited by the organoselenium chemical ethaselen (BBSKE). Through the formation of two covalent connections with the cysteine-497 and selenocystine-498 residues, BBSKE particularly targets the C-terminal redox core of TrxR [38]. BBSKE’s suppression of TrxR1 causes cells to produce more ROS due to inhibition of scavenging [39]. When taken with other anticancer drugs such sodium selenite [40] and the multi-targeted tyrosine kinase inhibitor sunitinib [39], BBSKE has demonstrated a synergistic effect. According to a recent publication, TrxR1 is inhibited by BBSKE, or the enzyme is knocked down, in cancer cells, which lowers the amount of PD-L1 expression [41]. Nevertheless, prior to BBSKE exposure, a therapy with the antioxidant N-acetylcysteine restored PD-L1 expression.
Similarly, the antipsychotic medicine trifluoperazine (TFP), which is also taken into consideration for the treatment of cancer [42][43][42,43], deserves to be included as another ROS-inducing substance. Recent research has shown that TFP increases ROS levels in colorectal cancer cells while also raising PD-L1 expression in these cancer cells and PD-1 expression in CD4+ and CD8+ T cells that infiltrate tumours [42].
Metformin and phenformin, two biguanide medications used to treat diabetes, have also demonstrated anticancer activity both in vitro and in vivo. They both promote oxidative stress-mediated apoptosis in cancer cells and inhibit PD-L1 expression, mainly via the Hippo signalling pathway [44][45][46][47][44,45,46,47]. They also both enhance the production of ROS. It has been shown that metformin promotes the interaction and phosphorylation of PD-L1 by the AMP-activated protein kinase (AMPK) protein, leading to its abnormal glycosylation and subsequent destruction [48][49][48,49]. They appear to increase the anticancer activity of PD-1 inhibition, not diminish it, and hence they do not reduce the efficacy of anti-PD-1 therapy [50]. Immune checkpoint inhibitors combined with metformin were given to subjects affected by melanoma or lung cancer [51][52][51,52]. By suppressing myeloid-derived suppressor cells and lowering tumour cell oxygen consumption, these drugs might amplify PD-1 blocking. Additionally, this would diminish intratumoural hypoxia [53].
The catechin by-product EGCG (epigallocatechin-3-gallate), which is prevalent in green tea, can likewise lessen intracellular ROS production and stop the loss of antioxidants. This natural substance can reduce the OS brought on by several stimuli, including, for instance, arsenic and cigarette smoke [54][55][56][54,55,56]. It is a powerful immune–epigenetic modulator for the treatment and/or prevention of cancer [57][58][57,58] and has a variety of targets, comprising histone deacetylases and metalloproteinases. Its anti-oxidative and free radical scavenging properties have received a lot of attention [59]. It is interesting to note that PD-L1 expression was shown to be decreased by EGCG in pulmonary tumour cell lines, and that PD-L1 suppression by EGCG led to a recovery of T cell function [60].
The action of aryl hydrocarbon receptor (AhR) appears to be more complex [61]. Initially, AhR was identified as a transcription factor controlling xenobiotic response [62]. As a result of ligand interaction, AhR is translocated into the nucleus where it heterodimerizes with Aryl hydrocarbon Receptor Nuclear Translocator (ARNT) to create an active transcription complex. In the absence of a ligand, AhR aggregates in the cytoplasm with other chaperone proteins. The cytochrome P450 enzymes, which include CYP1A1, CYP1A2, and CYP1B1, are a subfamily of metabolizing enzymes that are recognized by the AhR/ARNT complex as xenobiotic-responsive elements (XREs) [63]. In addition to controlling immunological checkpoint protein expression, AhR is essential in modulating immune tolerance and immune suppression [64]. For instance, AhR is involved in PD-1 and PD-L1 transcriptional activation [65][66][65,66]. Another enzyme implicated in immunological suppression, indoleamine 2′ 3′-dioxygenase 1 (IDO1), is transactivated by AhR as well [67].
Tryptophan (Trp) is an amino acid that IDO1 uses to enzymatically convert into kynurenine (Kyn), an oncometabolite that can decrease T cell cytotoxicity in the tumour microenvironment by depleting Trp [68]. A study revealed that the hydrogen peroxide-treated human keratinocyte cell line (HaCaT) had increased AhR activity, resulting in increased expression of its downstream targets, including cytochrome P450 genes. Intriguingly, AhR activation and its downstream signalling were increased by preincubating the whole culture media with hydrogen peroxide. The oxidant causes the synthesis of oxindole, a tryptophan catabolic product, according to a later mass spectrometric investigation. The fact that 2-oxindole can activate AhR was also demonstrated by the authors, strongly indicating that ROS may have a considerable influence on AhR signalling [69][70][69,70]. AhR’s role as an oncogene is mostly explained by the ROS accumulation caused by enhanced CYP activities, which favours malignant transformation by causing severe OS and increased DNA damage [71]. High AhR expression in breast cancer cells is significantly correlated with ROS build-up, which causes AhR to translocate into the nucleus and enhances its transcriptional activity [72][73][72,73].
In any case, although immune checkpoint inhibitors have transformed the way that cancer is treated, only a small percentage of patients get long-lasting improvements. Therefore, it is crucial to comprehend the connections between ROS and the PD-(L)1 checkpoint, particularly to aid in the development of novel medication combinations. Yet, as demonstrated above, the relationship between PD-L1 presence and ROS generation is complicated. ROS have the ability to either increase or decrease the amount of PD-L1 in cancer cells. PD-L1 expression is frequently encouraged by increased ROS production, and vice versa, ROS scavenging can inhibit PD-L1. In spite of that, there are clear exceptions when it comes to medications that boost ROS production while lowering PD-L1 expression and vice versa. Although no clear and distinct association can be inferred, drugs that alter ROS generation can have a considerable impact on PD-L1 expression [74] (Figure 3).
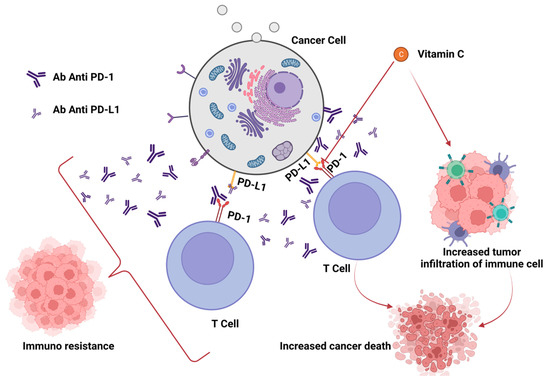
Figure 3. Effects of vitamin C on efficacy of checkpoint blockade treatment.
For instance, Auronafin and disulfiram are two ROS-producing medications that have been shown in studies to increase PD-L1 expression in cancer cells, whereas other ROS-enhancing compounds (such as ethaselen, chaetocin, and metformin) tend to lower PD-L1 expression. PD-L1 expression is increased in cells treated with As2O3 and disulfiram, but ROS scavenging is shown simultaneously with PD-L1 downregulation. However, in many instances (after cell treatment with the plant extract Anoectochilus formosanus), ROS production and PD-L1 expression showed a similar pattern. It has been discovered that human oncoviruses such as the EBV, which infect human primary monocytes to significantly increase the expression of PD-L1 on their surface by producing ROS, follow a similar pattern [75].
The impact of ROS-inducing drugs on the expression of PD-L1 is undoubtedly more complicated than has previously been documented, and these chemicals may also have additional modes of action. For instance, an alternative mechanism used by AhR could be the excess kynurenine produced by malignant cells and the impact of kynurenine on TME immune cells. Kynurenine may increase the expression of PD-L1 on G-MDSC, macrophages, and DCs, all of which express AhR, just as it does in cancerous cells. Through direct transcriptional regulation, AhR activation also produces the immunosuppressive CD39 ectoenzyme on macrophages and T cells. Furthermore, melanomas’ production of AhR and “transcellular” Kynurenine has been connected to PD-1 expression on CD8+ T cells in the TME. Chronic IFN production appears likely to aggravate each of these AhR-mediated consequences. It was surprising to learn that a large portion of the effect of IFN on Cd274 transcription in the MOC1 model is due to the AhR. It was also remarkable that the recognized IFN-induced induction of Ido was mostly AhR controlled. Although there have been suggestions that IFN and AhR signalling interact, it has been reported that AhR control of IFN-driven outcomes, notably PD-L1 and IDO activation, can be demonstrated in the context of cancer [65].
The condition of the check points and the redox balance in distinct neoplasms will be discussed in more detail in the sections that follow, as well as how changing OS might affect how well check point inhibitors prevent tumour growth in particular tumour pathologies.