Two-dimensional silicon carbide (2D SiC) is ma de of a single/few atomic layer of silicon carbide. or few layers of Si and C atoms. 2D SiC has a graphene-like honeycomb structure consisting of alternating Si and C atoms. In the monolayer SiC, the C and Si atoms bond through sp2 hybridization to form the SiC sheet.
As a direct wide bandgap semiconducting material, 2D SiC has the potential to bring revolutionary advances into opower etoelectronics, opto and electronic and other SiC-based dedevices. It can overcome current limitations with silicon, bulk SiC, and gapless graphene. In addition to SiC, which is the most stable form of monolayer silicon carbide, other compositions, i.e. SixCy, are also predicted to be energetically favorable. Depending on the stoichiometry and bonding, monolayer SixCy may behave as a semiconductor, semimetal or topological insulator. With different Si/C ratios, the emerging 2D silicon carbide materials could attain novel electronic, optical, magnetic, mechanical, and chemical properties that go beyond those of graphene, silicene, and already discovered 2D semiconducting materials.
- silicon carbide
- two-dimensional materials
- semiconductor
- optoelectronics
1. Introduction
The discovery of monolayer silicon carbide will accelerate various technological innovations in the posemiconductor sector. t-Moore era. As a wide bandgap semiconducting material with high thermal capability, SiC is a leading material for high-power electronics and high- temperature applications. However, due to quantum confinement and surface effects, 2D SiC offers tremendous unprecedented properties, which are absent in bulk SiC materials [1][2][3][4][5][1–5].
Unlike graphene, which is a pure one atom carbon material, 2D silicon carbide is a heteroatomic material that may exist in a variety of compositions and hence structures i.e.SixCy ,among others. Further, unlike graphene which can be exfoliated from bulk graphite via mechanical exfoliation, the synthesis of single-layer SiC is one of the most challenging and tricky syntheses among 2D materials, demanding a deep understanding of the atomic structure of the bulk SiC and its crystal structures. The main challenge is that bulk SiC is not a layered van der Waals material. It is a covalently bonded material with sp3 bonding between carbon and silicon along the c axis. As such, the formation of a monolayer silicon carbide requires phase transformation from sp3 to sp2. These structural challenges lead to the following fundamental questions: How will hexagonal 2D SiC be isolated from the tetrahedrally coordinated bulk SiC if the top-down approach would be adopted? Additionally, how easy would be the phase transformation from sp3 to sp2? Or at what thickness does the transformation take place? Furthermore, how stable is 2D SiC? Does it have an ideal planar structure or slightly buckled form? And more importantly is 2D SiC stable in air? Or is it highly reactive?
Herein, we address these outstanding questions by reviewing the latest efforts and progress in the field of 2D silicon carbide, focusing on the structure, properties, and potential applications of these emerging 2D materials. This paper is organized as follows. The first section will provide a fundamental understating of the structure of 2D silicon carbide. Then, the key properties of 2D SiC will be discussed. Finally, we will outline future opportunities and challenges.
2. The Structure of 2D Silicon Carbide
Structurally, 2D SiC is predicted to have a graphene-like honeycomb structure consisting of alternating Si and C atoms. In the monolayer SiC, the carbon and silicon atoms will bond through sp2 hybrid orbitals to form the SiC sheet, Figure 1. Various research groups have investigated the stability of planar 2D SiC, and all these studies have confirmed that 2D SiC is energetically stable and has a 100% planar structure with inherent dynamic stability [6][7][8][6–8]. They found that, while there may be a competition between sp2 hybridization preferred by C in its planar form and sp3 preferred by Si, the ground state of 2D SiC is completely flat, as planar 2D SiC has the lowest energy [9][10][11][9–11].
The predicted planarity feature is very important, and it contributes significantly to the development of several unprecedented properties. In fact, except graphene and hexagonal boron nitride, h-BN, most of the explored 2D materials, do not have stable planar structures. Instead, they stabilize their monolayer structures via i.e., a mix of bonding i.e., buckling. For instance, buckling values of 0.44 Å, 0.65 Å and 2.3 Å have been reported for silicene, germanene, and black phosphorous (BP), respectively [12][13][14][15][11–15]. In the case of silicene, it was even suggested that one approach to reducing buckling level is to use alternate atoms instead of pure silicon. Thus, given that 2D SiC could be considered as a heteroatomic form of silicene, it is reasonable that it has a stable planar structure.
As shown in Figure 1e, side view of silicene, as a result of mixed hybridization, sp3 and sp2, the bonds between adjacent atoms of the silicene lattice are buckled, resulting in a layer that is not completely flat. It is of note to mention that like graphite, black phosphorous is a van der Waals layered material. The buckling character affects the properties of 2D buckled materials significantly. For instance, because silicene does not have a perfect planar structure, it has a lower intrinsic electron/hole mobility than graphene.
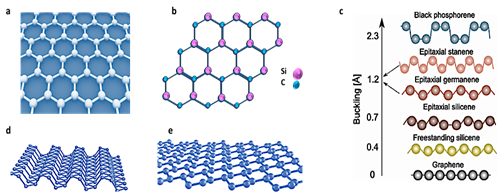
Figure 1. Schematics of crystal structure of graphene (a) and 2D SiC (b). Reported buckling for different 2D materials. (c) Unlike silicene and black phosphorous (BP), 2D SiC is 100% planar. (c) is reproduced from ref [15]. Atomic Structure of 2D black phosphorus (d). Credit: Institute for Basic Science. Atomic structure of silicene (e). Reprinted by permission from Nature [16], Copyright (2015).
Successful application of any 2D materials depends firstly on the material’s chemical and environmental stability. For instance, 2D BP-based devices have yet to be realized, because 2D BP suffers from a lack of environmental stability [17] [17]. In the case of silicon carbide monolayer, there has been some skepticism about the stability of planar monolayer SiC due to the high reactivity of Si=C bonding. Although this reasoning is understandable, since sp2 bonding is not the preferred configuration for bulk SiC, the case at ultralow thickness is different. The stability of the sp2 hybrid in monolayer SiC can be attained in several potential ways.
In fact, 2D SiC is not the first structure that has Si=C double bonding, as such the idea of planar 2D SiC is not without foundation. A variety of Si=C containing compounds, known as “silenes’’ have been reported in the past [18][19][20][18–20]. The stability of Si=C bond in such materials is attributed, to a great extent, to the depolarization of the Si=C double bond as a result of the electronic effects of the substituents on the double bond. Such substitutes reduce the natural polarity of the Si=C bond through effects on both the positive charge density at silicon, , and negative charge density, , at carbon [21][22][21,22]. In addition to the reduced polarity, steric protection due to bulky substituents, and aromaticity conjugation may also contribute to the stabilization of bonding between Si and C [23][24][25][26][27][20,23–27].
Similarly, the stability of monolayer 2D SiC can be discussed in terms of electronic effects and surface depolarization. Freeman et al. [28] used density functional theory (DFT) calculations to study phase transformation and stability in ultrathin wurtzite SiC, ZnO, GaN, BeO, and AIN films. They predicted that when wurtzite SiC or ZnO structures are thinned down to few atomic layers, they adopt graphitic like structure in which the atoms are threefold coordinated. This prediction was then confirmed experimentally for ultrathin films such as ZnO films [29], and AlN [30]. Uncompensated polarity and the transformation to graphitic-phase has also been reported for MgO ultrathin films [31][32][31,32]. Thus, although a significant gap of knowledge exists about the certain thickness at which phase transformation occurs, we do know that monolayer silicon carbide is stable and has a planar structure without any buckling. However, since bulk SiC has tetragonal bonding, a phase transformation from to is required as single layer SiC is isolated from bulk SiC. As a result of phase transformation, the length of the Si-C bonds gets reduced, from 1.89 to 1.79 Å, and the bond angle increases from 109 to 120 degrees. Depending on the configuration and stacking sequence, different values might be achieved for interlayer distances [33]. Additionally, the phonon dispersion of 2D SiC has also been investigated by various research groups [34][2,9,34], and the calculated phonon spectra show no imaginary frequencies, indicating the stability of 2D SiC. Table 1 summarizes the structural characteristics of 2D SiC vs. other related materials.
Table 1. Structural characteristics of 2D SiC and other related materials.
Material | Bond Length | (Å) | Lattice Constant | (Å) | Configuration | Interlayer Distance | (Å) | Refs | |||||||||||||||
2D SiC | 1.77-1.79 | 3.1 | planar | varies |
[2,10,35–41] |
||||||||||||||||||
6H-SiC | 1.89 | 3.08 | sp3 | 2.52 |
[36] |
[2,36] |
|||||||||||||||||
Graphene | 1.42 | 2.46 | planar | 1.42 |
[41] |
[2,41] |
|||||||||||||||||
Silicene | 2.27 | 3.8 | buckled | varies |
[42] |
[9,37,42] |
As a binary compound, 2D silicon carbide may exists in a variety of compositions i.e., 2D As such, thermodynamic and kinetic stability of 2D has been investigated by various theoretical studies [44][45][46][47][9,43–47]. Shi et al. used the cluster expansion method to investigate the stability of 2D honeycomb structures, Figure 2 [9]. As shown, all compositions have positive formation energies except , i.e., SiC, which has negative formation energy and lies below the straight lines connecting the two phases boundaries, x = 0 and 1. Negative formation energy is an indication of high stability. These findings agree very well with all other theoretical studies [43][48][6,43,48], confirming that SiC is the most stable stoichiometry in the whole Si−C system in its ground state. However, as shown, all other compositions have relatively small formation energies, less than 100 eV for some compounds, and negative cohesive energy, suggesting that these structures are not completely unstable. Instead, they are metastable, and perhaps could be stabilized in some ways in the future.
These results are very similar to the findings from bulk silicon carbide. In a related study [36], Hoffmann group studied the stability of phases at P=1 atm in bulk materials. They found that although the simplest stoichiometric composition ,i.e., SiC is the most stable phase, both Si3C and SiC3 are dynamically stable. Meaning although they are less stable than SiC, if they could be made, they would persist, as the activation energies for transforming them to SiC, C or Si is very large. More importantly, they found a relatively high stability of one type of graphene-like structure for SiC3 structures [36]. Interestingly, Zhao et al. [43] studied the stability of 2D SiC3, and 2D Si3C and similar results have been reported. Both 2D Si3C and 2D SiC3 are predicted to be topological insulators.
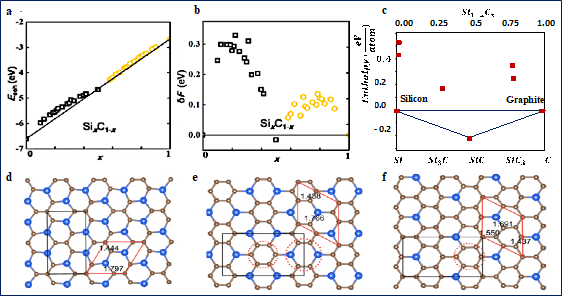
Figure 2. Cohesive energies, formation energies, and structures of SixCy materials. (a,b) Predicted cohesive energies and formation energies of 2D SixCy. Reprinted with permission from [9]. Copyright (2015) American Chemical Society. (c) predicated formation enthalpies of bulk . Reprinted with permission from [36]. Copyright (2013) American Chemical Society. As shown in (a), all investigated structures have negative cohesive energy, meaning that they are energetically favorable. These values also suggest that if these structures can be made, they will resist and will not break into graphene, or any other materials. However, as shown in b and c, only Si0.5C0.5 has a negative formation energy, an indication of high stability. (d–f) predicted structures for SiC2,SiC3,and SiC7 respectively. Republished from [49], with permission from IOP. As shown, unlike 2D SiC which has average bond length of 1.79 Å, two bond lengths exist in the presented structure, one belongs to C-C, and the other to Si-C. It should also be noted that more than a hundred structures have been predicted for 2D , and the presented structures here only represent a few examples of the most studied structures.
Additionally, it has been reported that carbon-rich compounds e.g., SiC2 or SiC3, are more stable than Si-rich compounds in their graphene-like structure. For example, using the global particle-swarm optimization algorithm and DFT calculations, Zhou et al. confirmed the thermodynamic stability of graphene-like SiC2 with an appropriate direct band gap [36]. In a similar study, Ref [50] studied and compared the stability of Si2C and SiC2 compounds and found that graphene-like Si2C sheet has a higher formation energy, and thus unstable. Similar to bulk material, Si-rich 2D composites e.g., Si2C and Si3C are more stable in a diamond like 4 coordinated structures [[50]36,50]. In addition to the structural differences, the key properties of 2D is determined by the Si/C stoichiometry as well. As a result of different composition, 2D silicon carbide could show a broad range of electronic and optical properties.
Regardless of the composition, alloying carbon and silicon atoms in such a planar two-dimensional binary system is very interesting, as this new 2D system offers a high level of capabilities and flexibilities, and functionalities, which are impossible in other closely related materials such as graphene or silicene. The next sections will provide a detailed discussion about the properties of 2D silicon carbide materials. First, electronic properties will be discussed.
3. Electronic Properties of 2D Silicon Carbide
Key properties of 2D SiC have been investigated using various density functional theory (DFT) approximations: Local-density approximations (LDA), generalized gradient approximations (GGA), GW corrections, Perdew, Burke, and Ernzerhof (PBE), and Heyd–Scuseria–Ernzerhof (HSE06) methods.
The electronic properties of 2D silicon carbide materials are determined basically through their electronic band structure. Figure 3 presents the band structures of hexagonal SiC monolayer, bulk 6H SiC, graphene, and silicene. As shown, both graphene and silicene are zero bandgap materials and in both materials, the valance and conduction bands, π and π*, meet at a single point at the Fermi level. However, hexagonal 2D SiC has a respectful band gap. The band gaps opening in 2D SiC is related to the electronegativity differences between Silicon and Carbon atoms, which would induce electron transfer from valance electrons of Si to the nearest C, so band gap emerges [51][34,41,51].
Theoretical calculations show that monolayer SiC is a direct bandgap semiconductor, that is in contrast with the indirect nature of the band gap in bulk SiC. Based on density functional theory, monolayer SiC has a direct band gap of 2.55 eV [52][53][54][55][37,52–55]. However, the calculated band gap increases to a higher value in the range of 3–4.8 eV when computed with GW quasiparticle corrections, GLLB-SC and other approximations [55][56][57][4,52,55–57]. The indirect-direct band gap transition characteristic in 2D SiC, is similar to the previously reported feature in other 2D materials such as 2D transition metal dichalcogenides (TMDs). This transition is attributed to the lack of any interlayer interactions in the TMDs monolayer [58]. It is of note to mention that TMDs are van der Waals layered materials like graphite. As such they can easily be fabricated via mechanical exfoliation.
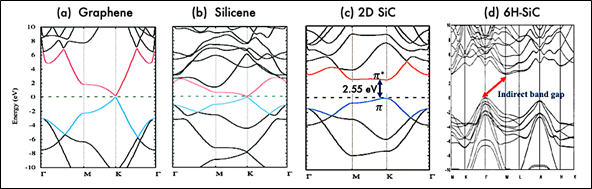
Figure 3. Electronic band structure of graphene, silicene, 2D SiC (a–c). Republished from [56] with permission from RSC. Electronic band structure of bulk 6H-SiC (d). Reproduced from [59] with permission from AIP publishing. Unlike graphene and silicene, 2D SiC have a band gap of about 2.55 eV (based on density functional theory (DFT)) due to its ionic nature. Further, 2D SiC has a direct band gap which is in contrast to indirect band gap in bulk SiC.
Electronic properties of 2D silicon carbide depend strongly on the number of layers, as well as the atomic ratio between carbon and silicon in Although there is a significant gap of knowledge about the electronic properties of few layer and multilayer SiC, the band structure of few layer SiC is expected to experience significant deviation from that of bulk SiC. Depending on the stacking sequences, e.g., AB, ABC, different band structures and thus, properties might be attained.
It was also reported that unlike monolayer SiC which has a direct bandgap, multilayer SiC has been found to have an indirect bandgap [60][61][37,60,61]. However, indirect-direct band gap crossover, similar to the case of MoS2 and other TMDs, is possible for few layer SiC. This crossover, which reaches its limit in monolayer SiC, is attributed to the reduced dimensionality and electronic confinement in the direction perpendicular to the c axis. The bandgap of few layer silicon carbide is expected to decrease as the number of layers increases. The latter can be attributed to the reduced dielectric screening in monolayer silicon carbide [62][58,62].
The size of the bandgap and carrier mobility in 2D showed a strong dependency on the atomic ratio between carbon and silicon [63][64][34,46,63,64]. Ref [9] used first-principles calculations combined with the cluster expansion method to study the structural and electronic properties of the monolayer . They found that most of the 2D materials exhibit semiconducting properties, while only two structures, and , are semimetallic, akin to graphene and silicene. Zhou et al. [50] reported that hexagonal SiC2 is a semiconductor with a direct band gap of 1.09 eV(HSE06) or 0.6 eV (PBE). Hexagonal SiC2 shows thermal stability up to 3000 K. Strained pentagonal 2D SiC2 is expected to have a very high hole mobility of , exceeding that of graphene [44]. Ref [43] used first-principles calculations combined with a tight-binding model to study the electronic properties of , and . They reported that both materials are topological insulators, and have Dirac cones centered at the K and K′ points.
The formation of Dirac cones is attributed to the preservation of the π-conjugate orbitals and hexagonal symmetry in both and structures as a result of their atomic arrangement. Similar to graphene, these materials might show the quantum-Hall effect. In a related study, Ref [63] used density functional calculations and particle-swarm optimization to investigate the electronic properties of . They reported that depending on the location of Si atoms, 2D may behave as a semiconducting or semimetal material. If Si atoms are located in a meta position, then sheet is a direct bandgap material. If silicon atoms are placed in a para position, then is a zero-bandgap semimetal with distorted Dirac cones.
Electronic properties of 2D and have also been investigated [46,47]. A bandgap of 0.73 eV, and high electron mobility of , along the [ direction, have been reported for [[47]47]. The carrier mobility of can be further increased via mechanical strain. On the other hand, it has been reported that graphene like is a semiconductor material with a direct band gap of 0.76 eV (PBE) or 1.13 eV (HSE06) [46]. Ref [65] studied electrical conductivity of SiC3 and SiC7. values of −1 and −1 have been reported for SiC3 at 200 K, and SiC7 at 600 K respectively. Here, is the electrical conductivity and is the time relaxations. Additionally, it has been reported that as the temperature increases, decreases for SiC3, and increases for SiC7.
The effects of the edge structure have also been investigated [66][54,66]. It has been reported that the armchair SiC nanoribbons are nonmagnetic semiconductors, while the zigzag nanoribbons are magnetic metals [56]. The band gap in armchair SiC nanoribbons can be tuned via hydrogen passivation. It was also reported that half-metallic zigzag edges can be turned metallic or semiconducting via functionalization with O or S atoms, respectively.
Additionally, the electronic properties of 2D SiC are highly affected by the defect level. For instance, local Dirac cones may form in 2D SiC, similar to graphene, as a results of C=C and Si=Si pairing [64]. C=C defects can be introduced into Si/C systems during the synthesis. In additional to defects, mechanical strain and chemical doping can also be used to tune the electronic properties of 2D SiC [67][68][69][60,67–69]. Not only the size of the bandgap, but even its nature can be changed from direct to indirect via mechanical strain [70]. Basically, since the charge distribution varies as the bond length changes, thus strain can play a critical role in tuning the electronic and optical properties of layered SiC [70].
As discussed, 2D silicon carbide materials, , benefit from highly tunable electronic properties. The band structure can be controlled via playing with Si/C composition, mechanical strain, and defects. This modifiability is of significant importance as it will enable the use of 2D silicon carbide for various applications.