The widely expressed G protein-coupled apelin receptor (APJ) is activated by two bioactive endogenous peptides, apelin and ELABELA (ELA). The apelin/ELA-APJ-related pathway has been found involved in the regulation of many physiological and pathological cardiovascular processes. Increasing studies are deepening the role of the APJ pathway in limiting hypertension and myocardial ischaemia, thus reducing cardiac fibrosis and adverse tissue remodelling, outlining APJ regulation as a potential therapeutic target for heart failure prevention.
- apelin
- ELABELA/Toddler/Apela
- APLNR/APJ receptor
- peptide analogue
- cardiovascular disease
- myocardial infarction
- heart failure
- hypertension
- angiogenesis
- fibrosis
1. Introduction
2. APJ and Its Endogenous Agonists
2.1. APJ
Apelinergic system history began in 1993 with the first description of the apelin receptor (APJ, also known as APLNR) based on its 31% homology with angiotensin II type 1 receptor (AT1R) [3]. Since the homologous sequences are primarily present in the seven hydrophobic transmembrane domains, APJ does not bind to angiotensin II (AngII) and for this reason was considered an orphan receptor for many years. At the same time, tissue distribution of APJ and AT1R is quite similar. APJ is widely expressed in the body and highly expressed mainly in the cardiovascular (CV) system where this receptor is present in cardiomyocytes (CMs), endothelial cells (ECs), and vascular smooth muscle cells (SMCs) [4][5][4,5]. The amino acid sequence of this receptor is made of 377 amino acid residues and is well conserved; in fact, the human APJ gene (APLNR) located on chromosome 11q12 displayed more than 90% sequence homology to murine APJ [6]. N-terminal glycosylation of APJ is essential for the proper receptor’s stability, protein folding, and binding to the ligand, while C-terminal palmitoylation favours the association to the cell membrane. Moreover, palmitoylation combined with phosphorylation allows APJ internalisation, dimerization, and the interaction with ligands [7]. APJ is a G-protein-coupled receptor (GPCR), and G proteins are formed by three different subunits (α, β, and γ) and are classified according to the α subunit in four families (i.e., Gαi/o, Gαq/11, Gαs, and Gα12/13) that trigger several different signalling pathways [8]. Upon C-terminal phosphorylation of APJ by GPCR kinases, β-arrestin is recruited and inhibits APJ activation by promoting its internalization [9]. It is also interesting to note that in the heart APJ may act as a mechanosensor for stretch through recruitment of β-arrestin [10]. The role of APJ resulted as pivotal in the cardiovascular system. During embryogenesis, APJ is expressed in mesoderm and APJ deficiency-caused defects in coronary vessel development [11][12][11,12]. In adults, APJ activation led to vasodilatation resulting in lowered blood pressure, angiogenesis, haemostasis, and anti-thrombotic effects. In the heart, it increases conduction velocity within CMs, has antiarrhythmic properties, and reduces myocardial hypertrophy and fibrosis [8]. APJ expression increases after ischaemia through the hypoxia-inducible factor-1 (HIF-1a) pathway [13].2.2. Apelin
Apelin was the first discovered natural endogenous ligand of APJ. Apelin was initially isolated from a cow’s stomach in 1998 by Tatemoto and co-workers [14], and afterwards, the presence of apelin mRNA has been detected in numerous tissues and organs, including adipose tissue, brain, liver, kidneys, skeletal muscles, heart, vessels, and lungs [15][16][17][15,16,17]. Apelin is encoded by a gene located on chromosome Xq 25–26 in various animal species including humans, mice, rats, and cows [16]. Apelin is initially produced as a preproprotein containing 77 amino acids, with the active sequence located in the C-terminal region. The removal of 22-unit signal peptide from N-terminal preproprotein generates the proprotein, also known as apelin-55 [18]. Endopeptidases further cleave apelin-55 into bioactive isoforms called apelin-36, -17, or -13, accordingly to the number of amino acid residues (Figure 1). Among them, the shorter peptides apelin-17, -13, and the pyroglutamate modified form of apelin-13 called [Pyr1]-apelin-13 have a higher affinity for APJ [14], while fragments shorter than 10 amino acids are biologically inactive [19]. Difference in isoform distribution was also observed; for example, [Pyr1]-apelin-13 is predominant isoform in the heart and plasma due to its resistance to degradation by peptidases [20][21][20,21], while apelin-36 is mainly detected in the lung, testis, and utero [22].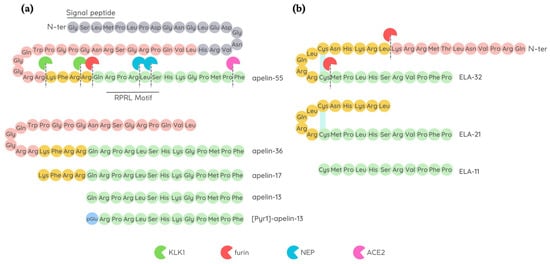
2.3. ELABELA
The discrepancy found in the foetal phenotypes between apelin knock-out and APJ-deficient mice, with observation of improper cardiac vessel formation and prenatal mortality, respectively, later led to the identification of Apela/ELABELA/Toddler (hereafter referred to as ELA) as a second APJ ligand [36][37][36,37]. It is encoded by the Apela/ELABELA/Toddler gene on chromosome 4q32.3, which is activated since the early embryonic stage. Apela gene encodes 54 amino acids pre-proprotein from which the active peptides ELA-32, -22, -21, and -11 (referring to the number of remaining amino acid residues at the C-terminus) are generated [36][38][36,38] (Figure 1). The C-terminal 13 residues of ELA are highly conserved across species and are required for APJ binding and the consequent activation of Gi protein alpha subunit (Gαi) as well as β-arrestin signalling pathways [38][39][38,39]. ELA-32 and ELA-11 differ in their membrane-interactive properties, providing a potential mechanism for distinctive signalling outcomes [40]. ELA is highly expressed in embryos, but is also present in some adult tissues, i.e., heart and blood vessels [32][41][42][32,41,42]. Like apelin, ELA displays a short half-life in plasma, and furins have been proposed as possible proteases able to cleave ELA-55 in two conserved di-arginine motifs [37][38][37,38]. The absence of the ELA gene causes early abnormalities in heart development, as observed in APJ-knockout animals [37], thus highlighting a crucial role of ELA in cardiac development and also leading to suggest a potential role of ELA in cardiac regeneration. In the rodent heart, ELA is predominantly expressed in the non-CM fraction, suggesting that ELA expression mainly occurs in fibroblasts and endothelial cells [42]. Paracrine and autocrine activities were suggested because of its low plasma level in humans (0.34 ± 0.03 nmol/L) [32], while in mouse heart, samples obtained 4 weeks after MI ELA expression resulted enhanced in the left ventricle of about 6 folds compared to control [42]. Due to the rather recent discovery of ELA, there is still much to find out about its endogenous isoform functions and their interaction with APJ.2.4. Physiological Ligand Activities in CV System
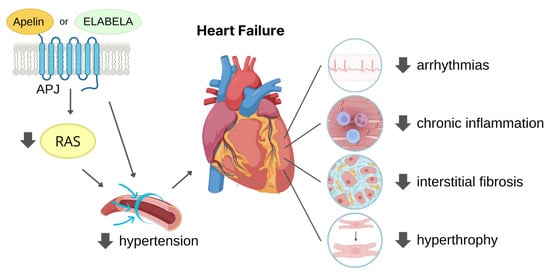
The apelin/ELA-APJ pathway is already involved in the embryonic heart development, even though the two endogenous ligands take part to this process at different time points or areas of the foetal heart as already reviewed by Kuba and co-authors [43]. In the adult CV system, upon binding to the APJ receptor, apelin and ELA exert mostly similar biological effects. After APJ binding, both apelin and ELA, at nanomolar concentration, exert positive inotropic effect on the in vivo rat hearts with comparable enhancement in the maximal rate of rise of left ventricular pressure (dP/dtmax), fractional shortening (c), heart rate, and cardiac output. Moreover, they reduce both left ventricular end diastolic pressure (LVEDP) and left ventricular end systolic pressure (LVESP), the latter likely due to a reduction of afterload (i.e., the resistance to cardiac output) caused by vasodilation [32][38][44][32,38,44]. Apelin expression appeared to be crucial for proper heart function as proved by the studies of Kuba and colleagues where the lack of apelin gene caused systolic dysfunction and the gradual decline in cardiac contractility, despite the absence of histological abnormalities [45]. Apelin-induced increase in contractility was extensively studied and showed the involvement of several pathways, among which the activation of pro-survival kinases PKC and ERK1/2 that favour the opening of sodium (NHE-1) and Ca2+ (NCX) channels [46][47][46,47]. Regarding ELA-dependent inotropic effects, ELA-32 was studied by Perjés and colleagues who attributed the increase in contractility to MEK1/2-ERK1/2 but not to PKC, whereas the apelin inotropic effect was mediated by both ERK1/2 and PKC [42]. However, the ELA-mediated inotropic effect was not completely suppressed after the inhibition of the MEK1/2–ERK1/2 pathway [42], thus suggesting that other additional pathways can be activated by this peptide. Further investigation will be needed to clarify this issue. Remarkably, in the healthy hearts, apelin treatment exerted a moderate inotropic effect that only lasted few minutes, while the inotropic effect resulted as particularly marked when administered in failing hearts [48][49][48,49]. More recently, a significant increase in lusitropic effect in response to apelin treatment was observed in mouse apelin knock-out CMs [50]. Apelin-induced positive lusitropic effect can be attributed to the preservation of SERCA activity avoiding the enhancement of intracellular calcium level, which may cause calcium overload-induced arrhythmogenesis [50][51][50,51]. Apelin and ELA are also vasoactive peptides with different mechanisms of action. Their administration results in a reduction in vascular resistance with consequent increase in blood flow accompanied by arterial pressure decrease. Apelin usually induces endothelial-induced vasodilation mediated by NO release via Akt activation [23][52][53][23,52,53], while in the case of endothelial disfunction, the peptide acts directly on smooth muscle cells triggering the opposite effect [54][55][54,55]. On the contrary, ELA-induced vasorelaxation did not require NO release [37]. Recently, the mechanisms of ELA-induced vasodilation have been elucidated by Sahinturk and collaborators who reported that endothelial-dependent vasorelaxation is mediated by prostanoids, while AMPK, PKC, and calcium-activated potassium channel activation was observed in the endothelial-independent pathway [56]. It is likely that beside the apelin/ELA-mediated increase in contractility, these peptides improved cardiac function by inducing vasodilation. In fact, the lowered afterload and the increased venous return (preload) allowed the heart to enhance the stroke volume. These APJ ligand effects can be useful in hearts with progressive function deterioration. Notably, both peptides are involved in the angiogenic process (see below). Moreover, their ability to attract and promote migration of endothelial cells led to consider apelin and ELA as chemotactic peptides in the angiogenic process [57][58][57,58]. APJ knockout mice exhibit abnormal body fluid balance [59] demonstrating the involvement of apelin and ELA in water homeostasis. Indeed, they reduce water intake, and apelin also increases water excretion [60][61][60,61]. Moreover, apelin also resulted as involved in energy metabolism by increasing glucose uptake by tissue cells and insulin sensitivity and mitochondrial bioactivity, as recently reviewed by Hu and collaborators [60].3. Cardioprotective Role of APJ Endogenous Ligands
In recent years, a growing body of evidence has highlighted the potential of apelin and ELA as therapeutic targets for the treatment of CV diseases, such as hypertension, ischaemic heart disease, and heart failure.3.1. Protection against Hypertension
Hypertension is a chronic medical condition in which the long-term high arterial blood pressure represents a relevant variable risk factor for cardiovascular disease morbidity and mortality because of increased incidence of MI and heart failure development [62][63][62,63]. Indeed, the persistence of high blood pressure induces LV hypertrophy and remodelling as adaptive response to cardiac output resistance [64]. The renin–angiotensin system (RAS) is a key pathway in the development and progression of hypertension. AngII, upon binding to AT1R, exerts a vasoconstrictor effect that plays a physiological role in arterial blood pressure control through the RAS. However, AngII–AT1R axis hyperactivation commonly leads to development and progression of hypertension [65]. AngII is formed starting from liver-produced angiotensinogen precursor which is cleaved by renin into Angiotensin I which in turn is converted to AngII through the catalytic activity of the angiotensin converting enzyme (ACE). Conversely, ACE2 negatively regulate RAS through the conversion of AngII into Ang(1–7), resulting in blood pressure reduction [66]. The apelin/ELA-APJ system can affect hypertension counteracting RAS (Figure 2).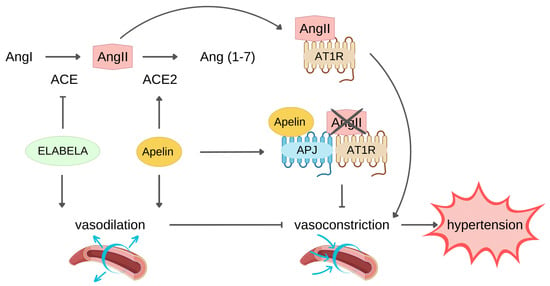
3.2. Protection against MI
MI is a critical cardiovascular event associated with high morbidity and mortality in which blood flow blockade prevents the O2 supply to the cardiac tissue. The restoration of blood flow, via thrombolytic therapy, primary percutaneous coronary intervention, and coronary artery bypass grafting (CABG), is essential for cardiac tissue salvage. However, after reperfusion, an exacerbation of myocardial damage takes place. For this reason, it is more correct talking of ischaemia-reperfusion (I/R) injury that consists of a series of events including cell death, inflammation, oxidative stress, calcium overload, and paradoxical pH that lead to cardiac tissue damage as well as impaired heart function (i.e., stunning of myocardium) [85]. Notably, it has been estimated that about 30% of patients develop I/R injury which seriously affected patient prognosis [85][86][85,86].4. APJ Peptide Agonists
Modified peptides can be designed to specifically bind a target receptor. Peptide modifications might modulate protein–protein interactions to improve target selectivity and affinity that play crucial roles during the persistent interaction between proteins. Furthermore, compared to small molecules and antibodies, modified peptides have the important advantage of causing a minor immune response [87][166]. The small size of APJ peptide ligands makes them easy to manipulate.4.1. APJ Endogenous Ligand Main Chain Modifications
The well-known short in vivo half-life that characterises both apelin and ELA poses a hurdle to their clinical translation. Many studies thus aim to develop more stable APJ ligands, based on either apelin or ELA peptide structure, through many different approaches, such as main-chain residue removal/addition, palmitoylation, and cleavage site modification of the several proteases involved in the degradation of parental peptides to obtain longer half-life in blood circulation as well as in tissues, trying to maintain at the same time apelin- or ELA-like cardiovascular effects (Table 1 and Table 2).|
APJ Ligand |
Substituted Residues |
||||
|---|---|---|---|---|---|
|
Cyclized Peptide |
Modified Residues |
Introduced Residues |
Cycle Position Effects |
Effects Ref |
|
Ref | |||||
|
[Pyr1]-apelin-13 |
Leu5, Ser 6 |
||||
Nπ-allyl-histidine linker | |||||
↑ binding affinity | |||||
↑ potency |
4.2. Antibody-Based/Bound APJ Agonists
|
Agonist |
Substituted Residues |
Introduced Residues |
Effects |
Ref |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
MM07 |
N-terminus |
||||||||||
|
JN241-9 |
between E104 and S105 to JN241 | - |
N-terminal cycle (MM07) |
↑ plasma protein stability |
tyrosine ↑ vasodilatation | ||||||
↑ cardiac function vs. [Pyr1]-apelin-13 |
agonist activity for cAMP β-arrestin recruitment |
[24] |
|||||||||
[ | ] | [186] |
Phe13 |
[(L-α-Me) Phe] |
↑ cardiac function ↓ hypertrophy |
↓ Pro12-Phe13 hydrolisis |
|||||
|
Macrocycle analogues | |||||||||||
|
MM202 (apelin-modified) |
Met |
Nle and Fluroaddition-C-terminal |
Pro12-Phe13 |
conformational constrained amino acids |
G-P-M−P-F ↑ binding affinity ↑ plasma protease stability |
[X-P-Nle-P-X] [B1-P-Nle-P-X] [B1-P-Nle-P-Xd] |
↑ plasma peptide half-life ↑ binding affinity vs. apelin-13 powerful modulators of cardiovascular system |
||||
|
C-terminal |
C-thermal exocyclic residues endocyclic residues |
↑ G-protein activation on over-arresting signalling binding affinity close to [Pyr1]-apelin-13 |
|||||||||
high binding affinity vs [Pyr1]-apelin-13 | = or ↑ potency |
than [Pyr1]-apelin-13 |
|||||||||
|
MM202-AlbudAb |
= or ↑ potency than [Pyr1]-apelin-13 | ||||||||||
|
Aia -Phe 1Nal-α,α-dibenzylglycine |
|||||||||||
|
MM202 and AlbudAb fusion |
↑ Gα12 pathways |
↑ cardiac function ↑ affinity and resistance to ACE2 cleavage ↑ in vitro and in vivo pharmacokinetics |
↓ heart rate | ||||||||
|
Phe13 |
Internal position | phenyl groups at the ortho-, meta-, and para-positions of Phe13 |
|||||||||
|
Fc-ELA-21 | slight effect on APJ binding affinity |
||||||||||
- |
Fc and ELA fusion |
↑ angiogenesis ↓ apoptosis ↑ cardiomyocyte proliferation ↓ heart fibrosis |
(L-α-Me) Phe or p-benzoyl-L-Phe (Bpa) |
β alanine spacer ↑ APJ functional selectivity for Gαi proteins no effect on cardiac contractility in healthy rats ↑ binding affinity vs. [Pyr1]-apelin-13 ↑ resistance to ACE2 cleavage ↓ hypertrophy and fibrosis |
[ |
Same APJ affinity of apelin-13 ↓ hypotensive effect similar to apelin-13 |
|||||
|
N-Teminus (pGlu) |
palmitic acid, (at Pro12) Aib, Nle |
↑ half-life (29 h in rat plasma) |
|||||||||
|
apelin-13′s His8 position | [ |
Nγ-allyl-Nγ-nosyl-α,γ-diamino-butanoic acid] |
|||||||||
|
N-Terminus |
Alkylated dipeptide |
↑ affinity in the low nM range |
|||||||||
|
extended and bulkier substituents |
binding affinity close to [Pyr1]-apelin-13 ↓ signalling potency for the β-arrestin 2 pathway |
||||||||||
|
C-Terminus |
positive charge |
binding affinity close to [Pyr1]-apelin-13 partial agonism for β-arrestin2 recruitment |
|||||||||
|
1NaI12 |
amino-indoloazepinone-Orn |
↑affinity for APJ ↑ Gαi1 activation, ↑ stability ↓ hypotensive effects |
|||||||||
|
apelin-12 |
N-terminus Met10 |
(NαMe) Arg Nle10 |
↑ proteolytic stability ↑ cardiac function ↓ membrane damage |
||||||||
|
apelin-17 |
Classical substituted |
P92 |
↑ plasma half-life sub-nanomolar affinity for APJ APJ internalization ↑ diuresis ↓ arterial blood pressure |
||||||||
|
LIT01-196 |
fluoroaddiction N-terminal |
||||||||||
|
↓ blood pressure in normotensive and hypertensive rats |
|||||||||||
|
Arg, Leu |
azapeptides |
↑ proteolytic stability vs. NEP |
|||||||||
|
l-hArg, l-Cha |
- |
↑ proteolytic stability vs. NEP prolonged lowering blood pressure effect in mice |
|||||||||
|
N-terminus |
palmitoylation PEGylation |
↑ plasma peptide half-life ↓ blood pressure in mice |
[30] |
||||||||
|
aromatic head groups carbonate analogues |
↑↑ proteolytic stability |
||||||||||
|
ELA |
Pro32 |
Tyr (OBn), Bpa, or 1Nal |
↑ binding affinity vs. ELA |
||||||||
|
N-terminus |
pyroglutamic acid (Pyr) residue to ELA(19-32) |
↓ arterial pressure ↓ inotropic effects on the heart ↑ half-life |
[38] |
4.3. Side Effect and Delivery Issues