Metabolic syndrome is characterized by abdominal obesity, high blood pressure, insulin resistance, a proinflammatory and prothrombotic state, atherogenic dyslipidemia , and low high-density lipoprotein cholesterol (HDL-C). Metabolic syndrom is mediated by oxidative stress and inflammation. Although the mechanisms are complicated and not very clear, this review elucidates the mechanisms and pathways that drive oxidative stress in contributing to metabolic syndrome., and low high-density lipoprotein cholesterol (HDL-C). Metabolic syndrom is mediated by oxidative stress and inflammation. Although the mechanisms are complicated and not very clear, this entry elucidates the mechanisms and pathways that drive oxidative stress in contributing to metabolic syndrome.
- oxidative stress
- inflammatory cytokines
- metabolic syndrome
- obesity
1. Introduction
2. Metabolic Syndrome Components
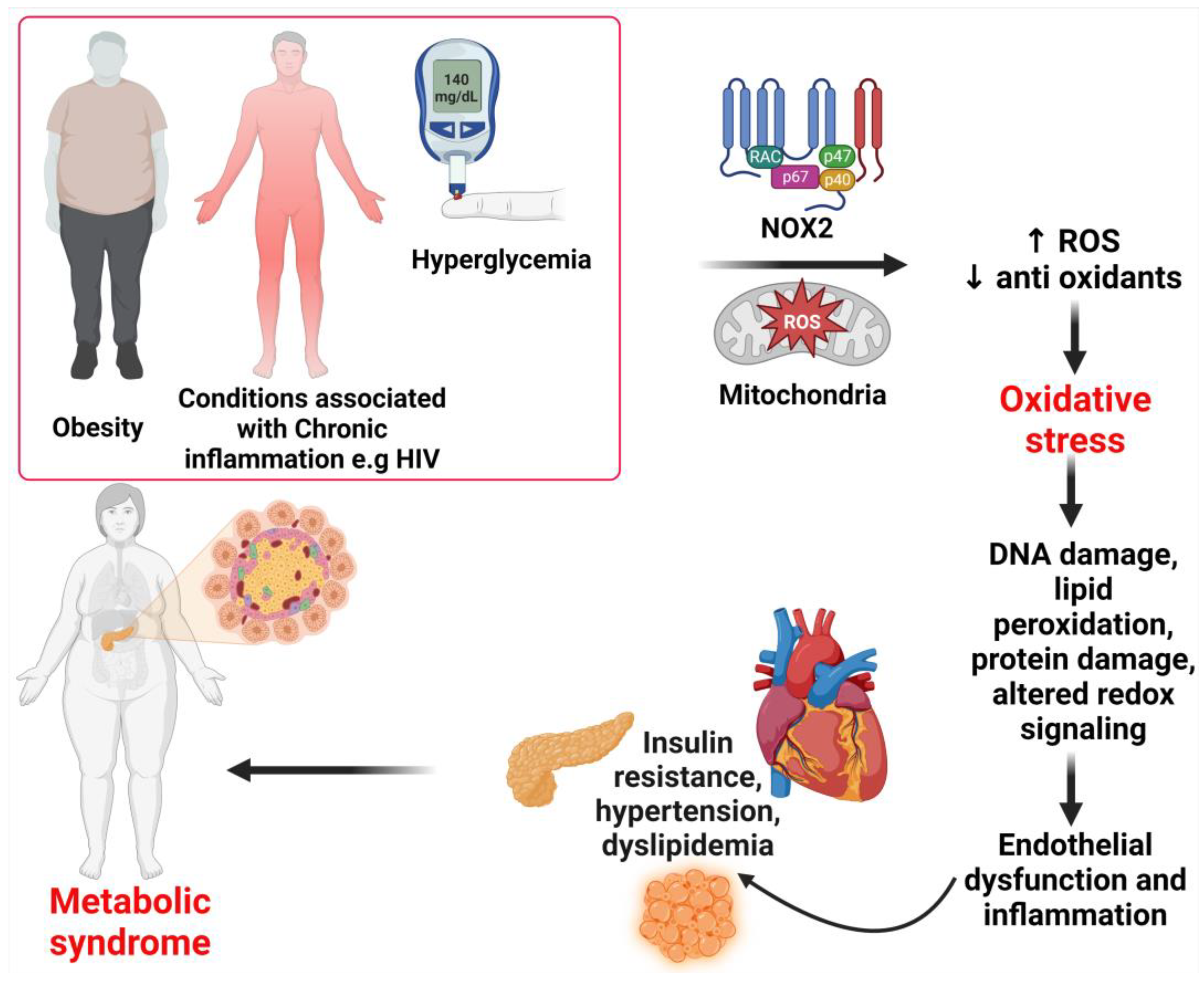
Metabolic syndrome is characterized by abdominal obesity, high blood pressure, insulin resistance (a risk factor for T2DM), a proinflammatory and prothrombotic state, and atherogenic dyslipidemia (high triglycerides, high apolipoprotein B, high low-density lipoprotein particle (LDL-p) number, and low high-density lipoprotein cholesterol (HDL-C) [3,5,13,14][3][5][13][14]. The components of metabolic syndrome are interrelated, as briefly described below. Obesity is the primary risk factor in the development of T2DM, and it is estimated that roughly 90 percent of people who have T2DM are either overweight or obese [15,16][15][16]. Obesity is also linked to an increased risk of CVD, which includes conditions such as high blood pressure, atherosclerosis, acute myocardial infarction, and heart failure [4]. Central obesity is defined as having an abdominal circumference that is greater than 102 cm for males and greater than 88 cm for women [17]. Central obesity is one of the most important factors in the etiology of metabolic syndrome, including insulin resistance [18]. Obesity is associated with low-grade inflammation, which may lead to insulin resistance, insulin deficiency, and metabolic disturbances [4]. Insulin, a peptide hormone released by pancreatic beta cells in response to rising blood glucose, blocks lipolysis, and hepatic gluconeogenesis, and increased glucose absorption in the liver, muscles, and adipose tissues [19]. Insulin resistance is an altered physiologic response to insulin stimulation of the target tissues, such as the liver, muscle, and adipose tissue, and the resistance hinders glucose metabolism, resulting in hypertrophy of beta cells, increased beta-cell insulin production, and hyperinsulinemia [20,21][20][21]. Insulin resistance may result in hyperglycemia, hypertension, dyslipidemia, visceral obesity, hyperuricemia, increased inflammatory markers, endothelial dysfunction, and thrombosis, which may lead to metabolic syndrome, nonalcoholic fatty liver disease (NAFLD), and T2DM through several complex mechanisms [21,22,23][21][22][23]. Lipid metabolism is critical to the etiology of insulin resistance and the subsequent development of metabolic syndrome [24]. Lipid changes also contribute to the diagnostic criteria for metabolic syndrome, and the two major lipids include fasting triglyceridemia >150 mg/dL and HDL cholesterol concentration <40 mg/dL. This lipid derangement is characterized by an increase in the synthesis of very low-density lipoproteins (VLDL), a decrease in the plasma’s lipolytic capacity, and an increased cholesterol ester transfer protein activity [18]. Patients with metabolic syndrome also exhibit hemostatic changes that can elevate the risk of both atherothrombotic and thromboembolic cardiovascular disease [18,25][18][25]. The atherothrombotic and thromboembolic changes result from endothelial dysfunction, which may be caused by chronic inflammation, dyslipidemia, and hypertension [18,25][18][25].3. Mechanisms of Reactive Oxygen Species and Their Role in the Development and Progression of Metabolic Syndrome
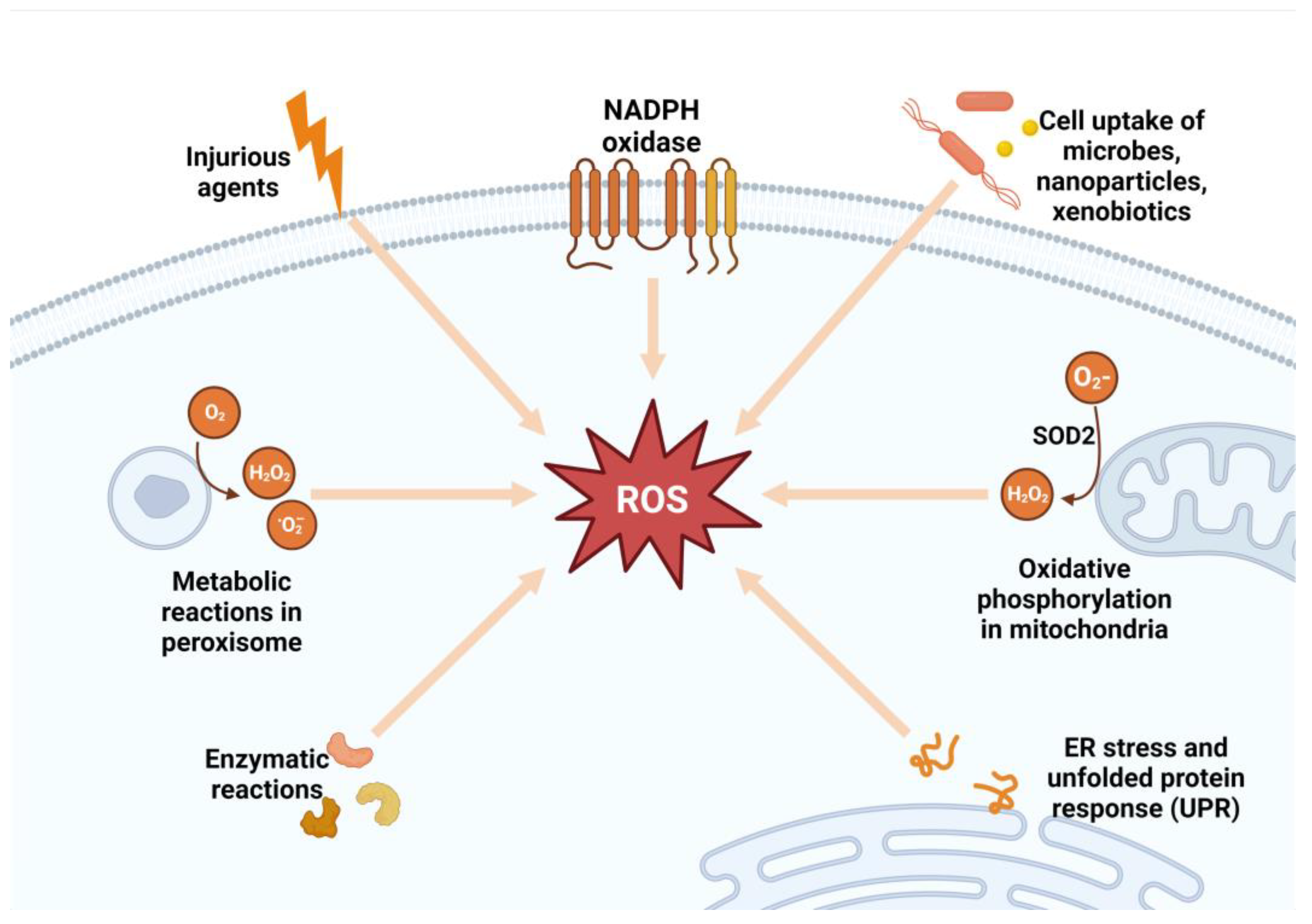
ROS production is tightly regulated by redox signaling and sensing mechanisms [26] (Figure 1).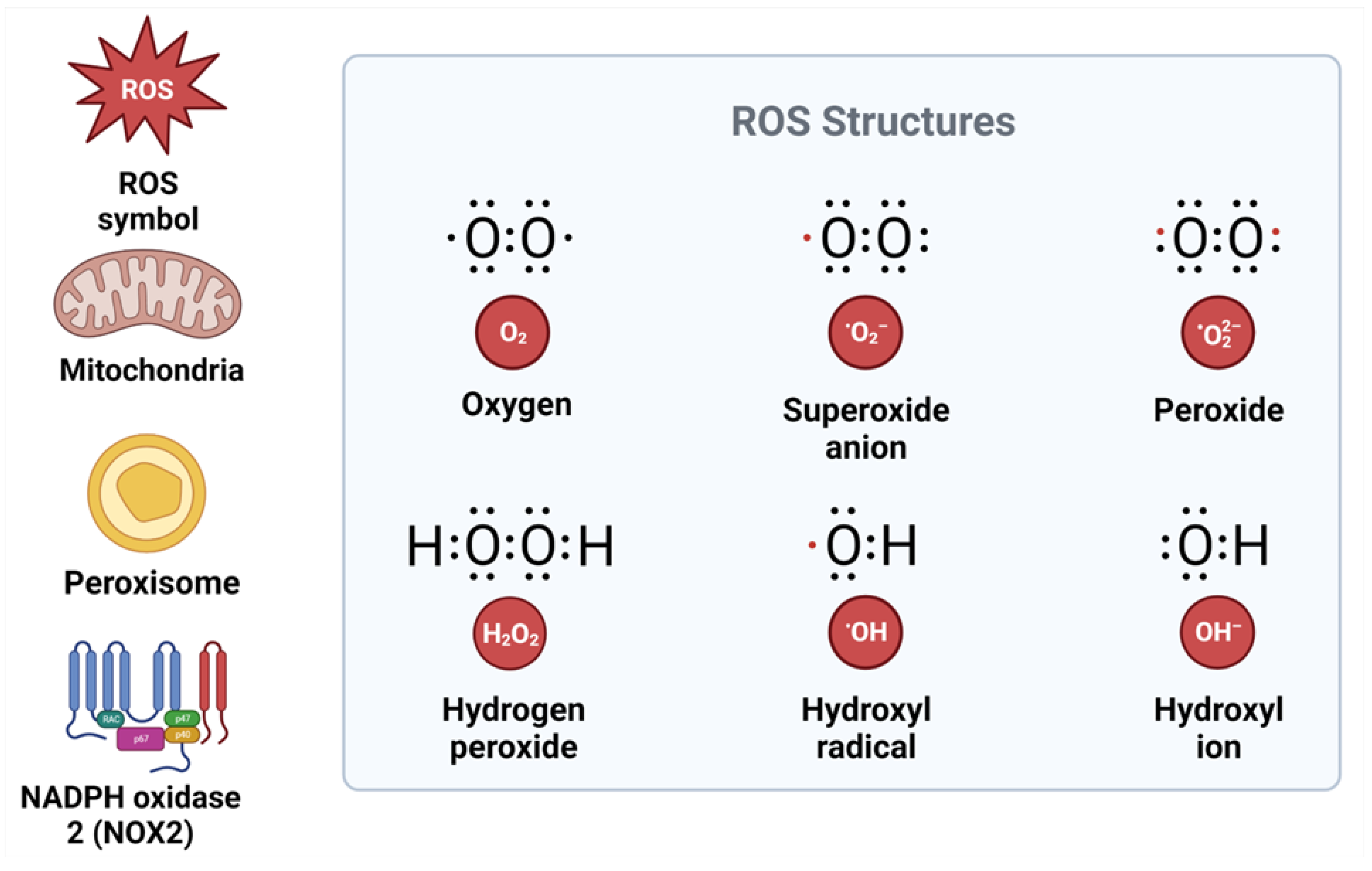
References
- Rochlani, Y.; Pothineni, N.V.; Kovelamudi, S.; Mehta, J.L. Metabolic Syndrome: Pathophysiology, Management, and Modulation by Natural Compounds. Ther. Adv. Cardiovasc. Dis. 2017, 11, 215–225.
- Gallardo-Alfaro, L.; Bibiloni, M.d.M.; Mascaró, C.M.; Montemayor, S.; Ruiz-Canela, M.; Salas-Salvadó, J.; Corella, D.; Fitó, M.; Romaguera, D.; Vioque, J.; et al. Leisure-Time Physical Activity, Sedentary Behaviour and Diet Quality Are Associated with Metabolic Syndrome Severity: The PREDIMED-Plus Study. Nutrients 2020, 12, 1013.
- Wilkinson, M.J.; Manoogian, E.N.C.; Zadourian, A.; Lo, H.; Fakhouri, S.; Shoghi, A.; Wang, X.; Fleischer, J.G.; Navlakha, S.; Panda, S.; et al. Ten-Hour Time-Restricted Eating Reduces Weight, Blood Pressure, and Atherogenic Lipids in Patients with Metabolic Syndrome. Cell Metab. 2020, 31, 92–104.e5.
- Monserrat-Mesquida, M.; Quetglas-Llabrés, M.; Capó, X.; Bouzas, C.; Mateos, D.; Pons, A.; Tur, J.A.; Sureda, A. Metabolic Syndrome Is Associated with Oxidative Stress and Proinflammatory State. Antioxidants 2020, 9, 236.
- Dieli-Conwright, C.M.; Courneya, K.S.; Demark-Wahnefried, W.; Sami, N.; Lee, K.; Buchanan, T.A.; Spicer, D.V.; Tripathy, D.; Bernstein, L.; Mortimer, J.E. Effects of Aerobic and Resistance Exercise on Metabolic Syndrome, Sarcopenic Obesity, and Circulating Biomarkers in Overweight or Obese Survivors of Breast Cancer: A Randomized Controlled Trial. J. Clin. Oncol. 2018, 36, 875–883.
- Vona, R.; Gambardella, L.; Cittadini, C.; Straface, E.; Pietraforte, D. Biomarkers of Oxidative Stress in Metabolic Syndrome and Associated Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 8267234.
- Franco, C.; Sciatti, E.; Favero, G.; Bonomini, F.; Vizzardi, E.; Rezzani, R. Essential Hypertension and Oxidative Stress: Novel Future Perspectives. Int. J. Mol. Sci. 2022, 23, 14489.
- Sies, H. Oxidative Stress: A Concept in Redox Biology and Medicine. Redox Biol. 2015, 4, 180–183.
- Touyz, R.M.; Rios, F.J.; Alves-Lopes, R.; Neves, K.B.; Camargo, L.L.; Montezano, A.C. Oxidative Stress: A Unifying Paradigm in Hypertension. Can. J. Cardiol. 2020, 36, 659.
- Han, T.S.; Lean, M.E. A Clinical Perspective of Obesity, Metabolic Syndrome and Cardiovascular Disease. JRSM Cardiovasc. Dis. 2016, 5, 2048004016633371.
- Todowede, O.O.; Sartorius, B. Prevalence of Metabolic Syndrome, Discrete or Comorbid Diabetes and Hypertension in Sub-Saharan Africa among People Living with HIV versus HIV-Negative Populations: A Systematic Review and Meta-Analysis Protocol. BMJ Open 2017, 7, e016602.
- Kunduraci, Y.E.; Ozbek, H. Does the Energy Restriction Intermittent Fasting Diet Alleviate Metabolic Syndrome Biomarkers? A Randomized Controlled Trial. Nutrients 2020, 12, 3213.
- Jeong, J.; Suh, Y.J. Association between Serum Uric Acid and Metabolic Syndrome in Koreans. J. Korean Med. Sci. 2019, 34, e307.
- Thomas, M.S.; Huang, L.; Garcia, C.; Sakaki, J.R.; Blesso, C.N.; Chun, O.K.; Fernandez, M.L. The Effects of Eggs in a Plant-Based Diet on Oxidative Stress and Inflammation in Metabolic Syndrome. Nutrients 2022, 14, 2548.
- Bhupathiraju, S.N.; Hu, F.B. Epidemiology of Obesity and Diabetes and Their Cardiovascular Complications. Circ. Res. 2016, 118, 1723–1735.
- Grant, B.; Sandelson, M.; Agyemang-Prempeh, B.; Zalin, A. Managing Obesity in People with Type 2 Diabetes. Clin. Med. 2021, 21, e327–e331.
- Schroder, J.D.; Falqueto, H.; Mânica, A.; Zanini, D.; de Oliveira, T.; de Sá, C.A.; Cardoso, A.M.; Manfredi, L.H. Effects of Time-Restricted Feeding in Weight Loss, Metabolic Syndrome and Cardiovascular Risk in Obese Women. J. Transl. Med. 2021, 19, 3.
- Pedro-Botet, J.; Ascaso, J.F.; Barrios, V.; De la Sierra, A.; Escalada, J.; Millán, J.; Mostaza, J.M.; Pérez-Martínez, P.; Pintó, X.; Salas-Salvadó, J.; et al. COSMIC Project: Consensus on the Objectives of the Metabolic Syndrome in Clinic. Diabetes Metab. Syndr. Obes. Targets Ther. 2018, 11, 683–697.
- Fahed, G.; Aoun, L.; Bou Zerdan, M.; Allam, S.; Bou Zerdan, M.; Bouferraa, Y.; Assi, H.I. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. Int. J. Mol. Sci. 2022, 23, 786.
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223.
- Wolosowicz, M.; Prokopiuk, S.; Kaminski, T.W. Recent Advances in the Treatment of Insulin Resistance Targeting Molecular and Metabolic Pathways: Fighting a Losing Battle? Medicina 2022, 58, 472.
- Kaur, J. A Comprehensive Review on Metabolic Syndrome. Cardiol. Res. Pract. 2014, 2014, 943162.
- Marušić, M.; Paić, M.; Knobloch, M.; Liberati Pršo, A.-M. NAFLD, Insulin Resistance, and Diabetes Mellitus Type 2. Can. J. Gastroenterol. Hepatol. 2021, 2021, 6613827.
- Denisenko, Y.K.; Kytikova, O.Y.; Novgorodtseva, T.P.; Antonyuk, M.V.; Gvozdenko, T.A.; Kantur, T.A. Lipid-Induced Mechanisms of Metabolic Syndrome. J. Obes. 2020, 2020, 5762395.
- Otamas, A.; Grant, P.J.; Ajjan, R.A. Diabetes and Atherothrombosis: The Circadian Rhythm and Role of Melatonin in Vascular Protection. Diab. Vasc. Dis. Res. 2020, 17, 1479164120920582.
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462.
- Samson, S.L.; Garber, A.J. Metabolic Syndrome. Endocrinol. Metab. Clin. N. Am. 2014, 43, 1–23.
- Jakubczyk, K.; Dec, K.; Kałduńska, J.; Kawczuga, D.; Kochman, J.; Janda, K. Reactive Oxygen Species—Sources, Functions, Oxidative Damage. Pol. Merkur. Lek. Organ Pol. Tow. Lek. 2020, 48, 124–127.
- DeVallance, E.; Li, Y.; Jurczak, M.J.; Cifuentes-Pagano, E.; Pagano, P.J. The Role of NADPH Oxidases in the Etiology of Obesity and Metabolic Syndrome: Contribution of Individual Isoforms and Cell Biology. Antioxid. Redox Signal. 2019, 31, 687–709.
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive Oxygen Species (ROS) Homeostasis and Redox Regulation in Cellular Signaling. Cell Signal. 2012, 24, 981–990.
- Zhu, J.; Wu, F.; Yue, S.; Chen, C.; Song, S.; Wang, H.; Zhao, M. Functions of Reactive Oxygen Species in Apoptosis and Ganoderic Acid Biosynthesis in Ganoderma Lucidum. FEMS Microbiol. Lett. 2019, 366, fnaa015.
- Veith, A.; Moorthy, B. Role of Cytochrome P450s in the Generation and Metabolism of Reactive Oxygen Species. Curr. Opin. Toxicol. 2018, 7, 44–51.
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial Electron Transport Chain: Oxidative Phosphorylation, Oxidant Production, and Methods of Measurement. Redox Biol. 2020, 37, 101674.
- Beckhauser, T.F.; Francis-Oliveira, J.; De Pasquale, R. Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity. J. Exp. Neurosci. 2016, 10, 23–48.
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.-A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793.
- Chung, H.S.; Wang, S.-B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine Oxidative Post-Translational Modifications: Emerging Regulation in the Cardiovascular System. Circ. Res. 2013, 112, 382–392.
- Baba, S.P.; Bhatnagar, A. Role of thiols in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 133–139.
- Tomin, T.; Schittmayer, M.; Honeder, S.; Heininger, C.; Birner-Gruenberger, R. Irreversible Oxidative Post-Translational Modifications in Heart Disease. Expert Rev. Proteom. 2019, 16, 681–693.
- Zhang, H.; Xu, R.; Wang, Z. Contribution of Oxidative Stress to HIF-1-Mediated Profibrotic Changes during the Kidney Damage. Oxid. Med. Cell Longev. 2021, 2021, 6114132.
- Padgett, L.E.; Broniowska, K.A.; Hansen, P.A.; Corbett, J.A.; Tse, H.M. The Role of Reactive Oxygen Species and Proinflammatory Cytokines in Type 1 Diabetes Pathogenesis. Ann. N. Y. Acad. Sci. 2013, 1281, 16–35.
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167.
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902.
- Čolak, E.; Pap, D. The Role of Oxidative Stress in the Development of Obesity and Obesity-Related Metabolic Disorders. J. Med. Biochem. 2021, 40, 1–9.
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642.
- Gaschler, M.M.; Stockwell, B.R. Lipid Peroxidation in Cell Death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425.
- Bekkouche, L.; Bouchenak, M.; Malaisse, W.J.; Yahia, D.A. The Mediterranean Diet Adoption Improves Metabolic, Oxidative, and Inflammatory Abnormalities in Algerian Metabolic Syndrome Patients. Horm. Metab. Res. Horm. Stoffwechselforschung Horm. Metab. 2014, 46, 274–282.
- Scioli, M.G.; Storti, G.; D’Amico, F.; Rodríguez Guzmán, R.; Centofanti, F.; Doldo, E.; Céspedes Miranda, E.M.; Orlandi, A. Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. J. Clin. Med. 2020, 9, 1995.