Type 2 diabetes mellitus (T2DM) is a metabolic disorder that impairsin which insulin signaling is impaired from reaching its effectors. Mitochondrial dysfunction causesleads to various diseases, including cardiovascular diseases, metabolic disorders, and cancer. MThe mitochondria are highly dynamic andin adjusting their functions to matchaccording to cellular conditions through . The shape, morphology, distribution, and number of mitochondria reflect their function through various processes, collectively known as mitochondrial dynamics, whichincluding mitochondrial fusion, fission, biogenesis, transport, and mitophagy. These processes determine the overall mitochondrial health and vitality. DMore evidence supports the idea that dysregulated mitochondrial dynamics play essential roles in the pathophysiology of insulin resistance, obesity, and T2DM, including as well as imbalanced dmitochondrial dynamics found in T2DM.
- fission
- fusion
- metabolic disorders
- mitochondrial biogenesis
- mitochondrial dynamics
- type 2 diabetes mellitus
1. Introduction
2. Mitochondrial Dynamics: Biogenesis, Transport, Fusion, Fission, and Mitophagy
2.1. Mitochondrial Biogenesis
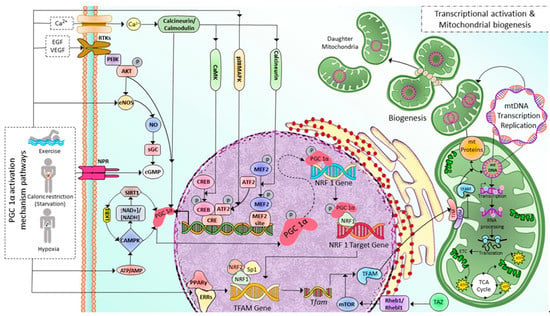
2.2. Mitochondrial Transport
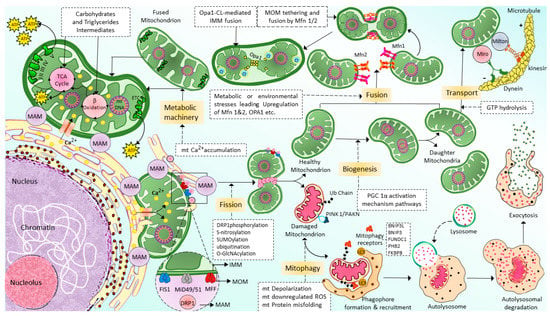
2.3. Mitochondrial Fusion
2.4. Mitochondrial Fission
2.5. Mitochondrial Selective Autophagy (Mitophagy)
3. Mitochondrial Dynamics in T2DM
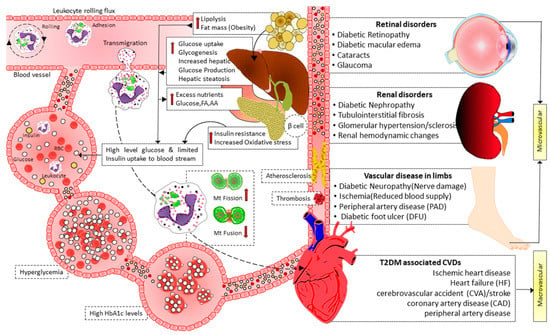
4. Dysregulated Mitochondrial Dynamics and Metabolism Play a Causal Role in Diabetes
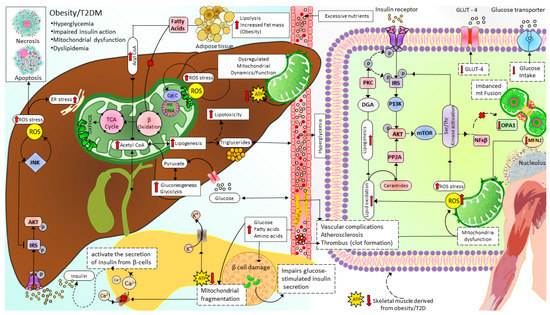
Mitochondrial dysfunction due to imbalanced mitochondrial dynamics leads to decreased OXPHOS, ATP production, β-oxidation of FFA [97][144], and enhanced ROS production [145]. Mitochondrial dysfunction plays a causal role by governing insulin secretion failure in β-cells and peripheral insulin resistance through the gluco-lipotoxicity [5][6] (Figure 4) or by impairing the regulation of glucose homeostasis in neurons of different cerebral regions [92][146][147][148][149][150] and glial cells in the brain (Figure 5). Excessive energy supply and low ATP demand increase mitochondrial fission, proton leak, and redox imbalance. On the other hand, in the depletion of nutrient supply and high ATP demand, the mitochondria are hyperfused, and respiration is optimized [151]. Accumulation of lipids/FFA increases diacylglycerol (DAG) and ceramides which activate protein kinase C (PKC) and protein phosphatase 2 (PP2A), respectively. PKC phosphorylates and inhibits insulin receptors. Meanwhile, PP2A dephosphorylates Akt and impairs insulin’s downstream signaling in which glucose transporter translocates to the cell membrane [152][153][154]. Furthermore, increased oxidative stress induces mitophagy and apoptosis, exacerbating the decrease in β-oxidation of FFA further, and ROS promotes Serine/threonine-specific protein kinases (Ser/Thr kinases) which phosphorylate and inhibit insulin receptor substrate (IRS) [155][156]. All these effects finally cause insulin resistance [157].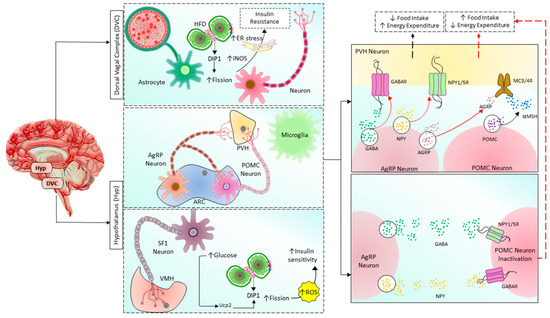
5. Therapeutic Opportunity of HDAC Inhibition for Mitochondrial Dysfunction in DM
Histone deacetylase (HDACs), one of the critical epigenetic regulators, has been shown to improve multiple metabolic disorders and regulate mitochondrial biogenesis and function. There are 18 mammalian HDAC isoforms that were identified and classified into four classes according to their sequence homology with yeast. The catalytic activity of classes I, II, and IV HDACs requires cofactor zinc ions, whereas class III HDACs are NAD+-dependent. Class I HDACs (HDAC1, HDAC2, HDAC3, and HDAC8) are ubiquitously distributed in the nucleus. Class II is further subdivided into class IIa (HDAC4, HDAC5, HDAC7, and HDAC9) and class IIb (HDAC6 and HDAC10). Class IIa HDACs shuttle between the cytoplasm and the nucleus with minimal catalytic activity. HDACs were recently identified as a potential therapeutic target for diabetes [222][223][224]. HDACs play important roles in regulating insulin-mediated cell signaling. Transcriptional expression of Glut4 is mainly regulated by myocyte enhancer factor 2 (MEF2), which binds to the Glut4 promoter. HDAC5 can interact with MEF2 and act as a transcriptional repressor of Glut4 by deacetylating histones and compacting chromatin structure. This inhibitory complex is released once HDAC5 is phosphorylated by AMPK and CaMK, allowing HDAC5 shuttle to cytoplasm and release from the inhibitory complex, instead, recruiting coactivator PGC-1α to increase Glut4 transcription. AMPK activator and HDAC5 siRNA are reported to enhance the transcriptional expression of Glut4 in myotube cells and adipocytes [225][226]. Moreover, HDAC2 can bind to insulin receptor substrate 1 (IRS1) in the liver cells of obese animals. HDAC2 inhibition with TSA (a pan HDAC inhibitor) or siRNA-mediated knockdown attenuates IRS-1 deacetylation and partially restores insulin signaling [227]. HDACs also regulate mitochondrial dynamics via deacetylating fission and fusion machinery and biogenesis via decreased expression of the primary regulator of MB, PGC-1a. The pan HDAC inhibitor SAHA induces mitochondrial elongation via decreasing expression of mitochondrial fission protein Fis1 and reduces the translocation of Drp1 to the mitochondria in Hep3B cells [228]. TSA also prevents mitochondrial fragmentation and provides neuroprotection against triggers for mitochondrial fragmentation (MPP+) in dopaminergic neurons via inhibiting histone deacetylation of the Mfn2 promoter and avoiding transcriptional repression of Mfn2 [229]. Additionally, SAHA can upregulate PGC-1 expression in mice and cultured cardiomyocytes [230]. Therefore, HDACs play pathological regulatory roles in glucose uptake, insulin resistance, and mitochondrial dynamics. Accordingly, HDACs may thus be a target for the treatment of insulin resistance and dysregulated mitochondrial dynamics. HDACs also regulate inflammation, oxidative stress, fibrosis, cell cycle and death, cellular metabolism, and other pathological processes through catalyzing deacetylation of the histones and non-histone proteins [227]. Accordingly, HDAC inhibition is expected to be a novel therapeutic strategy for DM through its effects on mitochondrial dynamics and several biological activities.6. Conclusions
Mitochondrial dynamics and metabolic disorders are commonly seen in overnutrition. Targeting mitochondrial dynamics (such as HDAC inhibition) via promoting mitochondrial biogenesis, restoration of mitochondrial fusion/fission balance, and mitophagy intervention are potential strategies for improving insulin resistance and T2DM. More clinical studies are needed to evaluate the efficacy of these interventions on human beings.References
- Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37 (Suppl. 1), S81–S90.
- Bullard, K.M.; Cowie, C.C.; Lessem, S.E.; Saydah, S.H.; Menke, A.; Geiss, L.S.; Orchard, T.J.; Rolka, D.B.; Imperatore, G. Prevalence of diagnosed diabetes in adults by diabetes type—United States, 2016. Morb. Mortal. Wkly. Rep. 2018, 67, 359.
- Xu, G.; Liu, B.; Sun, Y.; Du, Y.; Snetselaar, L.G.; Hu, F.B.; Bao, W. Prevalence of diagnosed type 1 and type 2 diabetes among US adults in 2016 and 2017: Population based study. BMJ 2018, 362, k1497.
- Archibald, J.M. Endosymbiosis and eukaryotic cell evolution. Curr. Biol. 2015, 25, R911–R921.
- Haastrup, M.O.; Vikramdeo, K.S.; Singh, S.; Singh, A.P.; Dasgupta, S. The Journey of Mitochondrial Protein Import and the Roadmap to Follow. Int. J. Mol. Sci. 2023, 24, 2479.
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta (BBA)-Bioenerg. 2017, 1858, 602–614.
- Dai, W.; Jiang, L. Dysregulated mitochondrial dynamics and metabolism in obesity, diabetes, and cancer. Front. Endocrinol. 2019, 10, 570.
- Archer, S.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251.
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287.
- Chen, H.; Chan, D.C. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 2017, 26, 39–48.
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884.
- Labbé, K.; Murley, A.; Nunnari, J. Determinants and functions of mitochondrial behavior. Annu. Rev. Cell Dev. Biol. 2014, 30, 357–391.
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646.
- Carelli, V.; Chan, D.C. Mitochondrial DNA: Impacting central and peripheral nervous systems. Neuron 2014, 84, 1126–1142.
- Lightowlers, R.N.; Taylor, R.W.; Turnbull, D.M. Mutations causing mitochondrial disease: What is new and what challenges remain? Science 2015, 349, 1494–1499.
- Kusminski, C.M.; Scherer, P.E. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol. Metab. 2012, 23, 435–443.
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312.
- Lahera, V.; de Las Heras, N.; López-Farré, A.; Manucha, W.; Ferder, L. Role of mitochondrial dysfunction in hypertension and obesity. Curr. Hypertens. Rep. 2017, 19, 11.
- Valera-Alberni, M.; Joffraud, M.; Miro-Blanch, J.; Capellades, J.; Junza, A.; Dayon, L.; Galindo, A.N.; Sanchez-Garcia, J.L.; Valsesia, A.; Cercillieux, A. Crosstalk between Drp1 phosphorylation sites during mitochondrial remodeling and their impact on metabolic adaptation. Cell Rep. 2021, 36, 109565.
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620.
- Suárez-Rivero, J.M.; Villanueva-Paz, M.; de la Cruz-Ojeda, P.; De la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; De Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Sánchez-Alcázar, J.A. Mitochondrial dynamics in mitochondrial diseases. Diseases 2016, 5, 1.
- Mullins, C. The Biogenesis of Cellular Organelles; Springer: Berlin/Heidelberg, Germany, 2005.
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 2007, 76, 701–722.
- Valero, T. Editorial (thematic issue: Mitochondrial biogenesis: Pharmacological approaches). Curr. Pharm. Des. 2014, 20, 5507–5509.
- Whitaker, R.M.; Corum, D.; Beeson, C.C.; Schnellmann, R.G. Mitochondrial biogenesis as a pharmacological target: A new approach to acute and chronic diseases. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 229–249.
- Sanchis-Gomar, F.; Luis Garcia-Gimenez, J.; Carmen Gomez-Cabrera, M.; Pallardo, F.V. Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches. Curr. Pharm. Des. 2014, 20, 5619–5633.
- Larsson, O.; Morita, M.; Topisirovic, I.; Alain, T.; Blouin, M.J.; Pollak, M.; Sonenberg, N. Distinct perturbation of the translatome by the antidiabetic drug metformin. Proc. Natl. Acad. Sci. USA 2012, 109, 8977–8982.
- Wood Dos Santos, T.; Cristina Pereira, Q.; Teixeira, L.; Gambero, A.; Villena, J.A.; Lima Ribeiro, M. Effects of Polyphenols on Thermogenesis and Mitochondrial Biogenesis. Int. J. Mol. Sci. 2018, 19, 2757.
- Marin, T.L.; Gongol, B.; Zhang, F.; Martin, M.; Johnson, D.A.; Xiao, H.; Wang, Y.; Subramaniam, S.; Chien, S.; Shyy, J.Y.-J. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci. Signal. 2017, 10, eaaf7478.
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab 2012, 23, 459–466.
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884s–890s.
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029.
- Marcinko, K.; Steinberg, G.R. The role of AMPK in controlling metabolism and mitochondrial biogenesis during exercise. Exp. Physiol. 2014, 99, 1581–1585.
- Dengler, F. Activation of AMPK under Hypoxia: Many Roads Leading to Rome. Int. J. Mol. Sci. 2020, 21, 2428.
- Chun, Y.; Kim, J. AMPK-mTOR Signaling and Cellular Adaptations in Hypoxia. Int. J. Mol. Sci. 2021, 22, 9765.
- Ren, Y.; Shen, H.M. Critical role of AMPK in redox regulation under glucose starvation. Redox Biol. 2019, 25, 101154.
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262.
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023.
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022.
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1. J. Biol. Chem. 2005, 280, 16456–16460.
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673.
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060.
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690.
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635.
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575.
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 1263–1268.
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47.
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566.
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698.
- Hwang, J.H.; Kim, K.M.; Oh, H.T.; Yoo, G.D.; Jeong, M.G.; Lee, H.; Park, J.; Jeong, K.; Kim, Y.K.; Ko, Y.G.; et al. TAZ links exercise to mitochondrial biogenesis via mitochondrial transcription factor A. Nat. Commun. 2022, 13, 653.
- Morita, M.; Gravel, S.P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711.
- Boldogh, I.R.; Pon, L.A. Interactions of mitochondria with the actin cytoskeleton. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2006, 1763, 450–462.
- Nangaku, M.; Sato-Yoshitake, R.; Okada, Y.; Noda, Y.; Takemura, R.; Yamazaki, H.; Hirokawa, N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell 1994, 79, 1209–1220.
- Tanaka, Y.; Kanai, Y.; Okada, Y.; Nonaka, S.; Takeda, S.; Harada, A.; Hirokawa, N. Targeted disruption of mouse conventional kinesin heavy chain kif5B, results in abnormal perinuclear clustering of mitochondria. Cell 1998, 93, 1147–1158.
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065.
- Griparic, L.; Van Der Wel, N.N.; Orozco, I.J.; Peters, P.J.; Van Der Bliek, A.M. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J. Biol. Chem. 2004, 279, 18792–18798.
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200.
- Chan, D.C. Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259.
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta (BBA)-Bioenerg. 2012, 1817, 1833–1838.
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192.
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383.
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99.
- Chen, K.H.; Dasgupta, A.; Lin, J.; Potus, F.; Bonnet, S.; Iremonger, J.; Fu, J.; Mewburn, J.; Wu, D.; Dunham-Snary, K.; et al. Epigenetic Dysregulation of the Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and Promotes Pulmonary Arterial Hypertension: Mechanistic and Therapeutic Implications. Circulation 2018, 138, 287–304.
- Kamerkar, S.C.; Kraus, F.; Sharpe, A.J.; Pucadyil, T.J.; Ryan, M.T. Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat. Commun. 2018, 9, 5239.
- Yu, R.; Jin, S.B.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748.
- Yu, R.; Liu, T.; Ning, C.; Tan, F.; Jin, S.-B.; Lendahl, U.; Zhao, J.; Nistér, M. The phosphorylation status of Ser-637 in dynamin-related protein 1 (Drp1) does not determine Drp1 recruitment to mitochondria. J. Biol. Chem. 2019, 294, 17262–17277.
- Civiletto, G.; Varanita, T.; Cerutti, R.; Gorletta, T.; Barbaro, S.; Marchet, S.; Lamperti, C.; Viscomi, C.; Scorrano, L.; Zeviani, M. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 2015, 21, 845–854.
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667.
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5.
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022.
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell Dev. Biol. 2020, 8, 467.
- Mao, K.; Wang, K.; Liu, X.; Klionsky, D.J. The scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev. Cell 2013, 26, 9–18.
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446.
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185.
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525.
- Gkikas, I.; Palikaras, K.; Tavernarakis, N. The role of mitophagy in innate immunity. Front. Immunol. 2018, 9, 1283.
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 2784–2790.
- Wu, H.; Chen, Q. Hypoxia activation of mitophagy and its role in disease pathogenesis. Antioxid. Redox Signal. 2015, 22, 1032–1046.
- Geng, G.; Liu, J.; Xu, C.; Pei, Y.; Chen, L.; Mu, C.; Wang, D.; Gao, J.; Li, Y.; Liang, J. Receptor-mediated mitophagy regulates EPO production and protects against renal anemia. Elife 2021, 10, e64480.
- Zuo, Z.; Jing, K.; Wu, H.; Wang, S.; Ye, L.; Li, Z.; Yang, C.; Pan, Q.; Liu, W.J.; Liu, H.-f. Mechanisms and functions of mitophagy and potential roles in renal disease. Front. Physiol. 2020, 11, 935.
- Sekine, S.; Youle, R.J. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2.
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550.
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119.
- Lu, S.; Liao, Z.; Lu, X.; Katschinski, D.M.; Mercola, M.; Chen, J.; Brown, J.H.; Molkentin, J.D.; Bossuyt, J.; Bers, D.M. Hyperglycemia Acutely Increases Cytosolic Reactive Oxygen Species via O-linked GlcNAcylation and CaMKII Activation in Mouse Ventricular Myocytes. Circ. Res. 2020, 126, e80–e96.
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070.
- King, G.L.; Loeken, M.R. Hyperglycemia-induced oxidative stress in diabetic complications. Histochem. Cell Biol. 2004, 122, 333–338.
- Hegarty, B.D.; Cooney, G.J.; Kraegen, E.W.; Furler, S.M. Increased efficiency of fatty acid uptake contributes to lipid accumulation in skeletal muscle of high fat-fed insulin-resistant rats. Diabetes 2002, 51, 1477–1484.
- Bastie, C.C.; Hajri, T.; Drover, V.A.; Grimaldi, P.A.; Abumrad, N.A. CD36 in myocytes channels fatty acids to a lipase-accessible triglyceride pool that is related to cell lipid and insulin responsiveness. Diabetes 2004, 53, 2209–2216.
- Bachmann, O.P.; Dahl, D.B.; Brechtel, K.; Machann, J.; Haap, M.; Maier, T.; Loviscach, M.; Stumvoll, M.; Claussen, C.D.; Schick, F. Effects of intravenous and dietary lipid challenge on intramyocellular lipid content and the relation with insulin sensitivity in humans. Diabetes 2001, 50, 2579–2584.
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800.
- Schrauwen, P.; Schrauwen-Hinderling, V.; Hoeks, J.; Hesselink, M.K. Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 266–271.
- Filippi, B.M.; Abraham, M.A.; Silva, P.N.; Rasti, M.; LaPierre, M.P.; Bauer, P.V.; Rocheleau, J.V.; Lam, T.K.T. Dynamin-Related Protein 1-Dependent Mitochondrial Fission Changes in the Dorsal Vagal Complex Regulate Insulin Action. Cell Rep. 2017, 18, 2301–2309.
- Serra, D.; Mera, P.; Malandrino, M.I.; Mir, J.F.; Herrero, L. Mitochondrial fatty acid oxidation in obesity. Antioxid. Redox Signal. 2013, 19, 269–284.
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 2004, 40, 185–194.
- Noeman, S.A.; Hamooda, H.E.; Baalash, A.A. Biochemical study of oxidative stress markers in the liver, kidney and heart of high fat diet induced obesity in rats. Diabetol. Metab. Syndr. 2011, 3, 17.
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658.
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950.
- Paltauf-Doburzynska, J.; Malli, R.; Graier, W.F. Hyperglycemic conditions affect shape and Ca2+ homeostasis of mitochondria in endothelial cells. J. Cardiovasc. Pharm. 2004, 44, 423–436.
- Vanhorebeek, I.; De Vos, R.; Mesotten, D.; Wouters, P.J.; De Wolf-Peeters, C.; Van den Berghe, G. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet 2005, 365, 53–59.
- Quirós, P.M.; Ramsay, A.J.; Sala, D.; Fernández-Vizarra, E.; Rodríguez, F.; Peinado, J.R.; Fernández-García, M.S.; Vega, J.A.; Enríquez, J.A.; Zorzano, A.; et al. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012, 31, 2117–2133.
- Sebastián, D.; Hernández-Alvarez, M.I.; Segalés, J.; Sorianello, E.; Muñoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528.
- Wang, L.; Ishihara, T.; Ibayashi, Y.; Tatsushima, K.; Setoyama, D.; Hanada, Y.; Takeichi, Y.; Sakamoto, S.; Yokota, S.; Mihara, K.; et al. Disruption of mitochondrial fission in the liver protects mice from diet-induced obesity and metabolic deterioration. Diabetologia 2015, 58, 2371–2380.
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645.
- Diaz-Morales, N.; Rovira-Llopis, S.; Bañuls, C.; Escribano-Lopez, I.; de Marañon, A.M.; Lopez-Domenech, S.; Orden, S.; Roldan-Torres, I.; Alvarez, A.; Veses, S.; et al. Are Mitochondrial Fusion and Fission Impaired in Leukocytes of Type 2 Diabetic Patients? Antioxid. Redox Signal. 2016, 25, 108–115.
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453.
- Finocchietto, P.; Perez, H.; Blanco, G.; Miksztowicz, V.; Marotte, C.; Morales, C.; Peralta, J.; Berg, G.; Poderoso, C.; Poderoso, J.J.; et al. Inhibition of mitochondrial fission by Drp-1 blockade by short-term leptin and Mdivi-1 treatment improves white adipose tissue abnormalities in obesity and diabetes. Pharm. Res. 2022, 178, 106028.
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319.
- Peng, L.; Men, X.; Zhang, W.; Wang, H.; Xu, S.; Fang, Q.; Liu, H.; Yang, W.; Lou, J. Involvement of dynamin-related protein 1 in free fatty acid-induced INS-1-derived cell apoptosis. PLoS ONE 2012, 7, e49258.
- Huang, M.; Wei, R.; Wang, Y.; Su, T.; Li, P.; Chen, X. The uremic toxin hippurate promotes endothelial dysfunction via the activation of Drp1-mediated mitochondrial fission. Redox Biol. 2018, 16, 303–313.
- Gao, L.; Liu, Y.; Wang, Y.; Chen, W.; Yang, K.; Li, J.; Lv, B.; Zhang, X.; Chi, J.; Liu, N.; et al. H2 relaxin ameliorates angiotensin II-induced endothelial dysfunction through inhibition of excessive mitochondrial fission. Biochem Biophys. Res. Commun. 2019, 512, 799–805.
- Zhang, B.; Guo, X.; Li, Y.; Peng, Q.; Gao, J.; Liu, B.; Wang, M. d-Chiro inositol ameliorates endothelial dysfunction via inhibition of oxidative stress and mitochondrial fission. Mol. Nutr. Food Res. 2017, 61, 1600710.
- Li, J.; Wang, Y.; Wang, Y.; Wen, X.; Ma, X.N.; Chen, W.; Huang, F.; Kou, J.; Qi, L.W.; Liu, B.; et al. Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. J. Mol. Cell. Cardiol. 2015, 86, 62–74.
- Li, Y.; Zhou, Z.H.; Chen, M.H.; Yang, J.; Leng, J.; Cao, G.S.; Xin, G.Z.; Liu, L.F.; Kou, J.P.; Liu, B.L.; et al. Inhibition of Mitochondrial Fission and NOX2 Expression Prevent NLRP3 Inflammasome Activation in the Endothelium: The Role of Corosolic Acid Action in the Amelioration of Endothelial Dysfunction. Antioxid. Redox Signal. 2016, 24, 893–908.
- Rogers, M.A.; Maldonado, N.; Hutcheson, J.D.; Goettsch, C.; Goto, S.; Yamada, I.; Faits, T.; Sesaki, H.; Aikawa, M.; Aikawa, E. Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ. Res. 2017, 121, 220–233.
- Ning, R.; Li, Y.; Du, Z.; Li, T.; Sun, Q.; Lin, L.; Xu, Q.; Duan, J.; Sun, Z. The mitochondria-targeted antioxidant MitoQ attenuated PM(2.5)-induced vascular fibrosis via regulating mitophagy. Redox Biol. 2021, 46, 102113.
- Abhijit, S.; Bhaskaran, R.; Narayanasamy, A.; Chakroborty, A.; Manickam, N.; Dixit, M.; Mohan, V.; Balasubramanyam, M. Hyperinsulinemia-induced vascular smooth muscle cell (VSMC) migration and proliferation is mediated by converging mechanisms of mitochondrial dysfunction and oxidative stress. Mol. Cell. Biochem. 2013, 373, 95–105.
- Zhang, X.; Chen, W.; Li, J.; Qi, S.; Hong, S.; Wang, Y.; Gao, L.; Shi, Z.; Liu, Y.; Liu, W.; et al. Involvement of mitochondrial fission in calcium sensing receptor-mediated vascular smooth muscle cells proliferation during hypertension. Biochem. Biophys. Res. Commun. 2018, 495, 454–460.
- Wu, Q.; Chen, Y.; Wang, Z.; Cai, X.; Che, Y.; Zheng, S.; Yuan, S.; Zhong, X. Mangiferin Inhibits PDGF-BB-Induced Proliferation and Migration of Rat Vascular Smooth Muscle Cells and Alleviates Neointimal Formation in Mice through the AMPK/Drp1 Axis. Oxid. Med. Cell. Longev. 2021, 2021, 3119953.
- Zhuang, X.; Maimaitijiang, A.; Li, Y.; Shi, H.; Jiang, X. Salidroside inhibits high-glucose induced proliferation of vascular smooth muscle cells via inhibiting mitochondrial fission and oxidative stress. Exp. Ther. Med. 2017, 14, 515–524.
- Wang, Q.; Zhang, M.; Torres, G.; Wu, S.; Ouyang, C.; Xie, Z.; Zou, M.-H. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes 2017, 66, 193–205.
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200.
- Zhong, Q.; Kowluru, R.A. Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8739–8746.
- Ding, M.; Dong, Q.; Liu, Z.; Liu, Z.; Qu, Y.; Li, X.; Huo, C.; Jia, X.; Fu, F.; Wang, X. Inhibition of dynamin-related protein 1 protects against myocardial ischemia–reperfusion injury in diabetic mice. Cardiovasc. Diabetol. 2017, 16, 19.
- Pegadraju, H.; Abby Thomas, J.; Kumar, R. Mechanistic and therapeutic role of Drp1 in the pathogenesis of stroke. Gene 2023, 855, 147130.
- Wu, Q.; Liu, J.; Mao, Z.; Tian, L.; Wang, N.; Wang, G.; Wang, Y.; Seto, S. Ligustilide attenuates ischemic stroke injury by promoting Drp1-mediated mitochondrial fission via activation of AMPK. Phytomedicine 2022, 95, 153884.
- Zhang, M.Y.; Zhu, L.; Zheng, X.; Xie, T.H.; Wang, W.; Zou, J.; Li, Y.; Li, H.Y.; Cai, J.; Gu, S.; et al. TGR5 Activation Ameliorates Mitochondrial Homeostasis via Regulating the PKCδ/Drp1-HK2 Signaling in Diabetic Retinopathy. Front. Cell Dev. Biol. 2021, 9, 759421.
- Zhang, M.Y.; Zhu, L.; Bao, X.; Xie, T.H.; Cai, J.; Zou, J.; Wang, W.; Gu, S.; Li, Y.; Li, H.Y.; et al. Inhibition of Drp1 ameliorates diabetic retinopathy by regulating mitochondrial homeostasis. Exp. Eye Res. 2022, 220, 109095.
- Chen, X.; Liang, J.; Bin, W.; Luo, H.; Yang, X. Anti-hyperlipidemic, Anti-inflammatory, and Ameliorative Effects of DRP1 Inhibition in Rats with Experimentally Induced Myocardial Infarction. Cardiovasc. Toxicol. 2021, 21, 1000–1011.
- Liu, D.; Zou, S.; Li, G.; Zhang, Q.; Chen, C.; Li, C.; Song, H.; Chen, S.; Wang, J.; Wu, Y.; et al. Downregulation of Uncoupling Protein 2(UCP2) Mediated by MicroRNA-762 Confers Cardioprotection and Participates in the Regulation of Dynamic Mitochondrial Homeostasis of Dynamin Related Protein1 (DRP1) After Myocardial Infarction in Mice. Front. Cardiovasc. Med. 2021, 8, 764064.
- Ding, J.; Zhang, Z.; Li, S.; Wang, W.; Du, T.; Fang, Q.; Wang, Y.; Wang, D.W. Mdivi-1 alleviates cardiac fibrosis post myocardial infarction at infarcted border zone, possibly via inhibition of Drp1-Activated mitochondrial fission and oxidative stress. Arch. Biochem. Biophys. 2022, 718, 109147.
- Xu, Y.; Wang, Y.; Wang, G.; Ye, X.; Zhang, J.; Cao, G.; Zhao, Y.; Gao, Z.; Zhang, Y.; Yu, B.; et al. YiQiFuMai Powder Injection Protects against Ischemic Stroke via Inhibiting Neuronal Apoptosis and PKCδ/Drp1-Mediated Excessive Mitochondrial Fission. Oxid. Med. Cell. Longev. 2017, 2017, 1832093.
- Kim, D.; Sesaki, H.; Roy, S. Reduced Levels of Drp1 Protect against Development of Retinal Vascular Lesions in Diabetic Retinopathy. Cells 2021, 10, 1379.
- Zhou, W.; Zhang, Y.; Jiao, Y.; Yin, W.; Dong, H.; Xu, S.; Tang, D.; Jiang, J.; Shao, J.; Wang, Z.; et al. Dexmedetomidine maintains blood-brain barrier integrity by inhibiting Drp1-related endothelial mitochondrial dysfunction in ischemic stroke. Acta Biochim. Biophys. Sin. (Shanghai) 2021, 53, 1177–1188.
- Chen, M.; Zhang, Q.; Wang, S.; Zheng, F. Inhibition of diabetes-induced Drp1 deSUMOylation prevents retinal vascular lesions associated with diabetic retinopathy. Exp. Eye Res. 2023, 226, 109334.
- Yang, D.Y.; Zhou, X.; Liu, Z.W.; Xu, X.Q.; Liu, C. LncRNA NEAT1 accelerates renal tubular epithelial cell damage by modulating mitophagy via miR-150-5p-DRP1 axis in diabetic nephropathy. Exp. Physiol. 2021, 106, 1631–1642.
- Chang, L.T.; Sun, C.K.; Wang, C.Y.; Youssef, A.A.; Wu, C.J.; Chua, S.; Yip, H.K. Downregulation of peroxisme proliferator activated receptor gamma co-activator 1alpha in diabetic rats. Int. Heart J. 2006, 47, 901–910.
- Zhang, Z.; Zhang, X.; Meng, L.; Gong, M.; Li, J.; Shi, W.; Qiu, J.; Yang, Y.; Zhao, J.; Suo, Y.; et al. Pioglitazone Inhibits Diabetes-Induced Atrial Mitochondrial Oxidative Stress and Improves Mitochondrial Biogenesis, Dynamics, and Function Through the PPAR-γ/PGC-1α Signaling Pathway. Front. Pharmcol. 2021, 12, 658362.
- Edwards, J.L.; Quattrini, A.; Lentz, S.I.; Figueroa-Romero, C.; Cerri, F.; Backus, C.; Hong, Y.; Feldman, E.L. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia 2010, 53, 160–169.
- Heinonen, S.; Buzkova, J.; Muniandy, M.; Kaksonen, R.; Ollikainen, M.; Ismail, K.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Vuolteenaho, K.; et al. Impaired Mitochondrial Biogenesis in Adipose Tissue in Acquired Obesity. Diabetes 2015, 64, 3135–3145.
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca2+ and AMPK/SIRT1. Nature 2010, 464, 1313–1319.
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124.
- Semple, R.K.; Crowley, V.C.; Sewter, C.P.; Laudes, M.; Christodoulides, C.; Considine, R.V.; Vidal-Puig, A.; O’Rahilly, S. Expression of the thermogenic nuclear hormone receptor coactivator PGC-1alpha is reduced in the adipose tissue of morbidly obese subjects. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 176–179.
- Roden, M. Muscle triglycerides and mitochondrial function: Possible mechanisms for the development of type 2 diabetes. Int. J. Obes. 2005, 29 (Suppl. S2), S111–S115.
- Kelley, D.E.; Goodpaster, B.; Wing, R.R.; Simoneau, J.A. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiol. 1999, 277, E1130–E1141.
- Simoneau, J.A.; Veerkamp, J.H.; Turcotte, L.P.; Kelley, D.E. Markers of capacity to utilize fatty acids in human skeletal muscle: Relation to insulin resistance and obesity and effects of weight loss. FASEB J. 1999, 13, 2051–2060.
- González-García, I.; Le Thuc, O.; Jastroch, M.; García-Cáceres, C. Divide et impera: How mitochondrial fission in astrocytes rules obesity. Mol. Metab. 2021, 45, 101159.
- Toda, C.; Kim, J.D.; Impellizzeri, D.; Cuzzocrea, S.; Liu, Z.W.; Diano, S. UCP2 Regulates Mitochondrial Fission and Ventromedial Nucleus Control of Glucose Responsiveness. Cell 2016, 164, 872–883.
- Ramírez, S.; Gómez-Valadés, A.G.; Schneeberger, M.; Varela, L.; Haddad-Tóvolli, R.; Altirriba, J.; Noguera, E.; Drougard, A.; Flores-Martínez, Á.; Imbernón, M.; et al. Mitochondrial Dynamics Mediated by Mitofusin 1 Is Required for POMC Neuron Glucose-Sensing and Insulin Release Control. Cell Metab. 2017, 25, 1390–1399.e1396.
- Dietrich, M.O.; Liu, Z.W.; Horvath, T.L. Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell 2013, 155, 188–199.
- Schneeberger, M.; Dietrich, M.O.; Sebastián, D.; Imbernón, M.; Castaño, C.; Garcia, A.; Esteban, Y.; Gonzalez-Franquesa, A.; Rodríguez, I.C.; Bortolozzi, A.; et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 2013, 155, 172–187.
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506.
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277.
- Bruce, C.R.; Risis, S.; Babb, J.R.; Yang, C.; Kowalski, G.M.; Selathurai, A.; Lee-Young, R.S.; Weir, J.M.; Yoshioka, K.; Takuwa, Y.; et al. Overexpression of sphingosine kinase 1 prevents ceramide accumulation and ameliorates muscle insulin resistance in high-fat diet-fed mice. Diabetes 2012, 61, 3148–3155.
- Schmitz-Peiffer, C.; Craig, D.L.; Biden, T.J. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J. Biol. Chem. 1999, 274, 24202–24210.
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15.
- Gutiérrez-Rodelo, C.; Roura-Guiberna, A.; Olivares-Reyes, J.A. Molecular Mechanisms of Insulin Resistance: An Update. Gac. Med. Mex. 2017, 153, 214–228.
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387.
- Komatsu, M.; Takei, M.; Ishii, H.; Sato, Y. Glucose-stimulated insulin secretion: A newer perspective. J. Diabetes Investig. 2013, 4, 511–516.
- Lin, H.-Y.; Weng, S.-W.; Chang, Y.-H.; Su, Y.-J.; Chang, C.-M.; Tsai, C.-J.; Shen, F.-C.; Chuang, J.-H.; Lin, T.-K.; Liou, C.-W.; et al. The Causal Role of Mitochondrial Dynamics in Regulating Insulin Resistance in Diabetes: Link through Mitochondrial Reactive Oxygen Species. Oxidative Med. Cell. Longev. 2018, 2018, 7514383.
- Chang, Y.H.; Lin, H.Y.; Shen, F.C.; Su, Y.J.; Chuang, J.H.; Lin, T.K.; Liou, C.W.; Lin, C.Y.; Weng, S.W.; Wang, P.W. The Causal Role of Mitochondrial Dynamics in Regulating Innate Immunity in Diabetes. Front. Endocrinol. 2020, 11, 445.
- Del Campo, A.; Parra, V.; Vásquez-Trincado, C.; Gutiérrez, T.; Morales, P.E.; López-Crisosto, C.; Bravo-Sagua, R.; Navarro-Marquez, M.F.; Verdejo, H.E.; Contreras-Ferrat, A.; et al. Mitochondrial fragmentation impairs insulin-dependent glucose uptake by modulating Akt activity through mitochondrial Ca2+ uptake. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1–E13.
- Dietrich, M.O.; Horvath, T.L. Feeding signals and brain circuitry. Eur. J. Neurosci. 2009, 30, 1688–1696.
- Broberger, C.; Johansen, J.; Johansson, C.; Schalling, M.; Hökfelt, T. The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc. Natl. Acad. Sci. USA 1998, 95, 15043–15048.
- Ollmann, M.M.; Wilson, B.D.; Yang, Y.K.; Kerns, J.A.; Chen, Y.; Gantz, I.; Barsh, G.S. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 1997, 278, 135–138.
- Horvath, T.L.; Bechmann, I.; Naftolin, F.; Kalra, S.P.; Leranth, C. Heterogeneity in the neuropeptide Y-containing neurons of the rat arcuate nucleus: GABAergic and non-GABAergic subpopulations. Brain Res. 1997, 756, 283–286.
- Diano, S. New aspects of melanocortin signaling: A role for PRCP in α-MSH degradation. Front. Neuroendocr. 2011, 32, 70–83.
- McClellan, K.M.; Calver, A.R.; Tobet, S.A. GABAB receptors role in cell migration and positioning within the ventromedial nucleus of the hypothalamus. Neuroscience 2008, 151, 1119–1131.
- Hahn, T.M.; Breininger, J.F.; Baskin, D.G.; Schwartz, M.W. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat. Neurosci. 1998, 1, 271–272.
- Kohno, D.; Sone, H.; Minokoshi, Y.; Yada, T. Ghrelin raises i via AMPK in hypothalamic arcuate nucleus NPY neurons. Biochem. Biophys. Res. Commun. 2008, 366, 388–392.
- Sternson, S.M.; Shepherd, G.M.; Friedman, J.M. Topographic mapping of VMH --> arcuate nucleus microcircuits and their reorganization by fasting. Nat. Neurosci. 2005, 8, 1356–1363.
- Takahashi, K.A.; Cone, R.D. Fasting induces a large, leptin-dependent increase in the intrinsic action potential frequency of orexigenic arcuate nucleus neuropeptide Y/Agouti-related protein neurons. Endocrinology 2005, 146, 1043–1047.
- Yang, Y.; Atasoy, D.; Su, H.H.; Sternson, S.M. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 2011, 146, 992–1003.
- Cone, R.D. Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 2005, 8, 571–578.
- Büch, T.R.; Heling, D.; Damm, E.; Gudermann, T.; Breit, A. Pertussis toxin-sensitive signaling of melanocortin-4 receptors in hypothalamic GT1-7 cells defines agouti-related protein as a biased agonist. J. Biol. Chem. 2009, 284, 26411–26420.
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdán, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484.
- Horvath, T.L.; Naftolin, F.; Kalra, S.P.; Leranth, C. Neuropeptide-Y innervation of beta-endorphin-containing cells in the rat mediobasal hypothalamus: A light and electron microscopic double immunostaining analysis. Endocrinology 1992, 131, 2461–2467.
- Beck, B. Neuropeptide Y in normal eating and in genetic and dietary-induced obesity. Philos. Trans. R. Soc. Lond B Biol. Sci. 2006, 361, 1159–1185.
- Kokot, F.; Ficek, R. Effects of neuropeptide Y on appetite. Min. Electrolyte Metab. 1999, 25, 303–305.
- Marraudino, M.; Bo, E.; Carlini, E.; Farinetti, A.; Ponti, G.; Zanella, I.; Di Lorenzo, D.; Panzica, G.C.; Gotti, S. Hypothalamic Expression of Neuropeptide Y (NPY) and Pro-OpioMelanoCortin (POMC) in Adult Male Mice Is Affected by Chronic Exposure to Endocrine Disruptors. Metabolites 2021, 11, 368.
- Spanswick, D.; Smith, M.A.; Groppi, V.E.; Logan, S.D.; Ashford, M.L. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature 1997, 390, 521–525.
- Spanswick, D.; Smith, M.A.; Mirshamsi, S.; Routh, V.H.; Ashford, M.L. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat. Neurosci. 2000, 3, 757–758.
- Van den Top, M.; Lee, K.; Whyment, A.D.; Blanks, A.M.; Spanswick, D. Orexigen-sensitive NPY/AgRP pacemaker neurons in the hypothalamic arcuate nucleus. Nat. Neurosci. 2004, 7, 493–494.
- Claret, M.; Smith, M.A.; Batterham, R.L.; Selman, C.; Choudhury, A.I.; Fryer, L.G.; Clements, M.; Al-Qassab, H.; Heffron, H.; Xu, A.W.; et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J. Clin. Investig. 2007, 117, 2325–2336.
- Cowley, M.A.; Smith, R.G.; Diano, S.; Tschöp, M.; Pronchuk, N.; Grove, K.L.; Strasburger, C.J.; Bidlingmaier, M.; Esterman, M.; Heiman, M.L.; et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003, 37, 649–661.
- Chen, S.R.; Chen, H.; Zhou, J.J.; Pradhan, G.; Sun, Y.; Pan, H.L.; Li, D.P. Ghrelin receptors mediate ghrelin-induced excitation of agouti-related protein/neuropeptide Y but not pro-opiomelanocortin neurons. J. Neurochem. 2017, 142, 512–520.
- Méquinion, M.; Foldi, C.J.; Andrews, Z.B. The Ghrelin-AgRP Neuron Nexus in Anorexia Nervosa: Implications for Metabolic and Behavioral Adaptations. Front. Nutr. 2019, 6, 190.
- Vong, L.; Ye, C.; Yang, Z.; Choi, B.; Chua, S., Jr.; Lowell, B.B. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 2011, 71, 142–154.
- Varela, L.; Horvath, T.L. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep. 2012, 13, 1079–1086.
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Münzberg, H.; Zhang, Z.Y.; Kahn, B.B.; et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 2015, 160, 88–104.
- Dodd, G.T.; Tiganis, T. Insulin action in the brain: Roles in energy and glucose homeostasis. J. Neuroendocr. 2017, 29, e2513.
- Timper, K.; Brüning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Model Mech. 2017, 10, 679–689.
- Shulman, R.G.; Rothman, D.L.; Behar, K.L.; Hyder, F. Energetic basis of brain activity: Implications for neuroimaging. Trends Neurosci. 2004, 27, 489–495.
- Nasrallah, C.M.; Horvath, T.L. Mitochondrial dynamics in the central regulation of metabolism. Nat. Rev. Endocrinol. 2014, 10, 650–658.
- Gómez-Valadés, A.G.; Pozo, M.; Varela, L.; Boudjadja, M.B.; Ramírez, S.; Chivite, I.; Eyre, E.; Haddad-Tóvolli, R.; Obri, A.; Milà-Guasch, M.; et al. Mitochondrial cristae-remodeling protein OPA1 in POMC neurons couples Ca(2+) homeostasis with adipose tissue lipolysis. Cell Metab. 2021, 33, 1820–1835.e1829.
- Santoro, A.; Campolo, M.; Liu, C.; Sesaki, H.; Meli, R.; Liu, Z.W.; Kim, J.D.; Diano, S. DRP1 Suppresses Leptin and Glucose Sensing of POMC Neurons. Cell Metab. 2017, 25, 647–660.
- Jin, S.; Diano, S. Mitochondrial Dynamics and Hypothalamic Regulation of Metabolism. Endocrinology 2018, 159, 3596–3604.
- Patel, B.; New, L.E.; Griffiths, J.C.; Deuchars, J.; Filippi, B.M. Inhibition of mitochondrial fission and iNOS in the dorsal vagal complex protects from overeating and weight gain. Mol. Metab. 2021, 43, 101123.
- Chang, C.R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39.
- Bo, T.; Yamamori, T.; Suzuki, M.; Sakai, Y.; Yamamoto, K.; Inanami, O. Calmodulin-dependent protein kinase II (CaMKII) mediates radiation-induced mitochondrial fission by regulating the phosphorylation of dynamin-related protein 1 (Drp1) at serine 616. Biochem. Biophys. Res. Commun. 2018, 495, 1601–1607.
- Kashatus, D.F.; Lim, K.-H.; Brady, D.C.; Pershing, N.L.; Cox, A.D.; Counter, C.M. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell Biol. 2011, 13, 1108–1115.
- Merz, K.E.; Hwang, J.; Zhou, C.; Veluthakal, R.; McCown, E.M.; Hamilton, A.; Oh, E.; Dai, W.; Fueger, P.T.; Jiang, L.; et al. Enrichment of the exocytosis protein STX4 in skeletal muscle remediates peripheral insulin resistance and alters mitochondrial dynamics via Drp1. Nat. Commun. 2022, 13, 424.
- Civitarese, A.E.; Carling, S.; Heilbronn, L.K.; Hulver, M.H.; Ukropcova, B.; Deutsch, W.A.; Smith, S.R.; Ravussin, E. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007, 4, e76.
- Mesquita, P.H.C.; Vann, C.G.; Phillips, S.M.; McKendry, J.; Young, K.C.; Kavazis, A.N.; Roberts, M.D. Skeletal Muscle Ribosome and Mitochondrial Biogenesis in Response to Different Exercise Training Modalities. Front. Physiol. 2021, 12, 725866.
- Viña, J.; Gomez-Cabrera, M.C.; Borras, C.; Froio, T.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Pallardo, F.V. Mitochondrial biogenesis in exercise and in ageing. Adv. Drug Deliv. Rev. 2009, 61, 1369–1374.
- Holloszy, J.O. Regulation of mitochondrial biogenesis and GLUT4 expression by exercise. Compr. Physiol. 2011, 1, 921–940.
- Hood, D.A.; Uguccioni, G.; Vainshtein, A.; D’Souza, D. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle: Implications for health and disease. Compr. Physiol. 2011, 1, 1119–1134.
- Pengam, M.; Amérand, A.; Simon, B.; Guernec, A.; Inizan, M.; Moisan, C. How do exercise training variables stimulate processes related to mitochondrial biogenesis in slow and fast trout muscle fibres? Exp. Physiol. 2021, 106, 938–957.
- Nishio, Y.; Kanazawa, A.; Nagai, Y.; Inagaki, H.; Kashiwagi, A. Regulation and role of the mitochondrial transcription factor in the diabetic rat heart. Ann. N. Y. Acad. Sci. 2004, 1011, 78–85.
- Hood, D.A. Invited Review: Contractile activity-induced mitochondrial biogenesis in skeletal muscle. J. Appl. Physiol (1985) 2001, 90, 1137–1157.
- Dillon, L.M.; Rebelo, A.P.; Moraes, C.T. The role of PGC-1 coactivators in aging skeletal muscle and heart. IUBMB Life 2012, 64, 231–241.
- Wenz, T.; Rossi, S.G.; Rotundo, R.L.; Spiegelman, B.M.; Moraes, C.T. Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. USA 2016, 113, E8502.
- Wei, P.; Guo, J.; Xue, W.; Zhao, Y.; Yang, J.; Wang, J. RNF34 modulates the mitochondrial biogenesis and exercise capacity in muscle and lipid metabolism through ubiquitination of PGC-1 in Drosophila. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 1038–1046.
- Egan, B.; Carson, B.P.; Garcia-Roves, P.M.; Chibalin, A.V.; Sarsfield, F.M.; Barron, N.; McCaffrey, N.; Moyna, N.M.; Zierath, J.R.; O’Gorman, D.J. Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor coactivator-1 mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J. Physiol. 2010, 588, 1779–1790.
- Edgett, B.A.; Foster, W.S.; Hankinson, P.B.; Simpson, C.A.; Little, J.P.; Graham, R.B.; Gurd, B.J. Dissociation of increases in PGC-1α and its regulators from exercise intensity and muscle activation following acute exercise. PLoS ONE 2013, 8, e71623.
- Song, K.; Zhang, Y.; Ga, Q.; Bai, Z.; Ge, R.L. Increased Insulin Sensitivity by High-Altitude Hypoxia in Mice with High-Fat Diet-Induced Obesity Is Associated with Activated AMPK Signaling and Subsequently Enhanced Mitochondrial Biogenesis in Skeletal Muscles. Obes. Facts 2020, 13, 455–472.
- Short, K.R.; Vittone, J.L.; Bigelow, M.L.; Proctor, D.N.; Rizza, R.A.; Coenen-Schimke, J.M.; Nair, K.S. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes 2003, 52, 1888–1896.
- Russell, A.P.; Feilchenfeldt, J.; Schreiber, S.; Praz, M.; Crettenand, A.; Gobelet, C.; Meier, C.A.; Bell, D.R.; Kralli, A.; Giacobino, J.P.; et al. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes 2003, 52, 2874–2881.
- Garnier, A.; Fortin, D.; Zoll, J.; N’Guessan, B.; Mettauer, B.; Lampert, E.; Veksler, V.; Ventura-Clapier, R. Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle. FASEB J. 2005, 19, 43–52.
- Baar, K.; Wende, A.R.; Jones, T.E.; Marison, M.; Nolte, L.A.; Chen, M.; Kelly, D.P.; Holloszy, J.O. Adaptations of skeletal muscle to exercise: Rapid increase in the transcriptional coactivator PGC-1. FASEB J. 2002, 16, 1879–1886.
- Pilegaard, H.; Saltin, B.; Neufer, P.D. Exercise induces Transient Transcriptional Activation of the PGC-1α Gene in Human Skeletal Muscle; Wiley Online Library: Hoboken, NJ, USA, 2003.
- He, F.; Huang, Y.; Song, Z.; Zhou, H.J.; Zhang, H.; Perry, R.J.; Shulman, G.I.; Min, W. Mitophagy-mediated adipose inflammation contributes to type 2 diabetes with hepatic insulin resistance. J. Exp. Med. 2021, 218, e20201416.
- Dewanjee, S.; Vallamkondu, J.; Kalra, R.S.; Chakraborty, P.; Gangopadhyay, M.; Sahu, R.; Medala, V.; John, A.; Reddy, P.H.; De Feo, V.; et al. The Emerging Role of HDACs: Pathology and Therapeutic Targets in Diabetes Mellitus. Cells 2021, 10, 1340.
- Christensen, D.P.; Dahllöf, M.; Lundh, M.; Rasmussen, D.N.; Nielsen, M.D.; Billestrup, N.; Grunnet, L.G.; Mandrup-Poulsen, T. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol. Med. 2011, 17, 378–390.
- Sharma, S.; Taliyan, R. Histone deacetylase inhibitors: Future therapeutics for insulin resistance and type 2 diabetes. Pharmacol. Res. 2016, 113, 320–326.
- McGee, S.L.; van Denderen, B.J.; Howlett, K.F.; Mollica, J.; Schertzer, J.D.; Kemp, B.E.; Hargreaves, M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 2008, 57, 860–867.
- Weems, J.; Olson, A.L. Class II histone deacetylases limit GLUT4 gene expression during adipocyte differentiation. J. Biol. Chem. 2011, 286, 460–468.
- Kaiser, C.; James, S.R. Acetylation of insulin receptor substrate-1 is permissive for tyrosine phosphorylation. BMC Biol. 2004, 2, 23.
- Lee, J.S.; Yoon, Y.G.; Yoo, S.H.; Jeong, N.Y.; Jeong, S.H.; Lee, S.Y.; Jung, D.-i.; Jeong, S.-Y.; Yoo, Y.H. Histone deacetylase inhibitors induce mitochondrial elongation. J. Cell. Physiol. 2012, 227, 2856–2869.
- Zhu, M.; Li, W.W.; Lu, C.Z. Histone deacetylase inhibitors prevent mitochondrial fragmentation and elicit early neuroprotection against MPP+. CNS Neurosci. Ther. 2014, 20, 308–316.
- Yang, J.; He, J.; Ismail, M.; Tweeten, S.; Zeng, F.; Gao, L.; Ballinger, S.; Young, M.; Prabhu, S.D.; Rowe, G.C.; et al. HDAC inhibition induces autophagy and mitochondrial biogenesis to maintain mitochondrial homeostasis during cardiac ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2019, 130, 36–48.