Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Xing-gang Mao and Version 2 by Sirius Huang.
Glioblastoma (GBM) is lack of effective treatment and the prognosis of GBM patients is still very poor despite accumulated progresses. Hypoxia is an essential factor for the initiation and progression of GBM, especially for the glioma stem like cells (GSCs). Hypoxia induced many target genes which form a complicated molecular interacting network, influencing a lot of tumor behaviors by regulating key signal pathways. In addition, hypoxia has great impact on the interplayed niches of GCSs.
- glioblastoma
- stem cell
- hypoxia
- niche
- autophagy
- therapy
- cancer stem cell
- molecular interacting network
- glioma
1. Introduction
Glioblastoma (GBM) is identified as one of the most malignant solid brain cancers, with an average annual incidence of about 4.45 per 100,000 population [1]. GBM patients present clinical manifestations of headache, weakness, vague vision, seizure and/or dizziness, depending on tumor location and degree of neurological impairment. The average diagnostic age of GBM patients is 64-years-old, with more females than males (6:4) [2]. With advancement of technology, novel imaging tools offer great assistance for oncologists in the diagnosis of GBM. Apart from conventional computed tomography (CT) and magnetic resonance imaging (MRI), dynamic contrast-enhanced (DCE) MRI, functional MRI, diffusion tensor imaging (DTI), magnetic resonance spectroscopy (MRS), diffusion-weighted imaging (DWI), single-photon emission computed tomography (SPECT), and positron emission tomography (PET) are all useful in clinical practice [3][4][3,4].
It has been proposed that GBMs have intrinsic cellular heterogeneity which consists of differentiated cells, quiescent cells, and glioblastoma stem cells (GSCs) [5]. Differentiated cells mainly contribute to tumorigenesis, proliferation, and invasion of glioblastoma. Quiescent cells are able to transdifferentiate into stem-like cells, and re-acquire self-renewal ability [6]. GSCs are recognized as reservoirs of tumor-initiating cells, accounting for therapeutic failure and GBM recurrence. The hypothesis of cancer stem cell (CSC) arises from human acute leukemia. Hematopoietic stem cells (HSCs) are a small population of multipotent cells with the potential of proliferation, self-renewal, differentiation, and regeneration of original tumors [7]. GSCs kept a certain degree of neural stem cell (NSC) features such as self-renewal and multi-differentiation potential [8]. NSC was first described in grown-up mammalians and mainly exists in two regions: one is the subventricular zone (SVZ) between the striatum and the lateral ventricle, and another is the subgranular zone (SGZ) within the dentate gyrus of the hippocampus [9][10][9,10].
Hypoxia plays a paramount important role in neuronal development which is a prerequisite for the neural crest cell migration [11]. Hypoxia mainly functions through hypoxia-inducible factors (HIFs, HIF1α, and HIF2α). In normoxia, HIF1α is hydroxylated and combined with a cancer suppressor Von Hippel-Lindau (VHL) to undergo ubiquitination process [12]. Under hypoxia, the HIF1α protein is speedily accumulated within cells and contributes to subsequent gene transactivation. HIF1α promotes glycolysis via upregulating critical enzymes of glycolytic pathway, such as HK2 (hexokinase 2) and pyruvate PDK1 (pyruvate dehydrogenase kinase 1). The literature has reported that HIF1α regulates stemness and differentiation of early NSC population via activating neural repressor Hes1 [13]. Suppression of HIF1α by meloxicam could exert antiproliferative efficacy in hepatocellular carcinoma (HCC) and lead to caspase-reliant apoptosis of HCC in hypoxia [14]. HIF2α is an intimate isoform of HIF1α [15]. Contrary to widespread expression of HIF1α in nearly all cells, HIF2α is selectively expressed in stem cells and endothelial cells of cancer. HIF1α shows high sensitivity towards oxygen concentration while HIF1β demonstrates constitutive expression regardless of oxygen concentration [16]. The HIF1α/HIF1β compound could translocate into the nucleus and further command genes that contain the hypoxia-response consensus sequence (HRE) [17].
Hypoxic areas in GBMs could be attributed to multiple factors such as upregulated cellular proliferation, insufficient oxygen diffusion, widespread tissue necrosis, broken blood-brain barrier, and aberrant tumor vascularization. Hypoxia is closely linked with the neoplastic biology of GBMs. Upregulated HIF expression in hypoxia promotes proliferation, infiltration, and self-renewal of GSC, ultimately leading to an enhanced level of therapeutic-resistance.
2. GSC and Hypoxia-Related Signatures
One of the central issues for studying GSCs is to identify the GSCs, primarily by using suitable molecular markers. It has been reported that CD9, CD133 (prominin-1), Olig2, integrin αβ, aldehyde dehydrogenase (ALDH), CD44, Sox2, Oct4, nestin, and the feature of side population (SP) can be used as signatures of GSCs [17][18][19][20][21][17,18,19,20,21] (Table 1). These markers are useful in the identification of stem cells, thus propelling relative studies. Via detecting expression of Oct4, Olig2, and nestin, it was reported that ING5 (a member of the epigenetic regulators ING family) could accelerate self-renewal of GSC, enhance its stem-cell pool, and block its lineage differentiation [22]. When hypoxia is presented, many of these GSC markers are upregulated. Seidle et al. observed that SP-related genes are upregulated in hypoxia in three adherent glioma cells [23].They also discovered that SP marker genes are highly expressed in both peri-vascular and hypoxic niches where both HIF1α and HIF2α are highly expressed [23]. However, whether all these cell markers can be applied precisely to identify stem cells remains controversial. Some CD133− cells also have the properties of GSCs and high plasticity of generating CD133+ cells. Currently, the gold criterion to determine GSCs remains the competence of recapitulating original parent tumors under the condition of orthotopical transplantation. Therefore, further investigations are necessary to uncover intrinsic GSC features.
Table 1.
GSC and hypoxia-regulated signatures, genes, and pathways.
| Signature | Gene | Pathways | lncRNA | Protein |
|---|---|---|---|---|
| CD9 | EGFR | DLK1 | lncRNA H19 | HILPDA (HIG2) |
| SP | TP53 mutation | Notch (CBF1) | ||
| CD133 | IDH mutation | VEGF | ||
| Olig2 | MCT4 | JAK1//2-STAT3 | ||
| integrin αβ | PP2A | Wnt (TCF-1, LEF-1) | ||
| ALDH | Klf4 | avβ8-integrin-TGF-β1 | ||
| CD44 | ABCB1 | |||
| Sox2 | PTEN | |||
| Oct4 | PML | |||
| nestin |
3. GSC and Hypoxia-Related Genes
GBMs can be further classified into four subtypes: mesenchymal, neural, proneural, and classical subtypes. Neurofibromin 1 (NF1) deletion, chromosome 7 enrichment, platelet-derived growth factor (PDGF) amplification, and tumor suppressor PTEN deficiency are discovered in these four types respectively [24]. In addition, p16 loss, epidermal growth factor receptor (EGFR) amplification, chromosome 22q loss, TP53 mutation, and CDKN2A loss are the most common and prominent signal alterations in GBMs [25] (Table 1). Among them, TP53 mutation is present in both primary and recurrent GBMs [26]. Recurrent GSC is able to accumulate temozolomide-associated mutations over primary GSC after chemo-therapy [26]. IDH1 is an oncogene which localizes in cytoplasm and peroxisome. IDH1 mutation is a symbol of early tumorigenesis, suppression of which could enhance sensitivity of GBM to chemotherapy. Glioma patients with IDH-mutation display better prognoses compared to those with IDH-wildtype. In the latest WHO classification, GBM only represents IDH-wildtype GBM, while IDH-mutant GBM, which was considered to account for 10% of GBMs in the past [27], is considered as a different subtype of diffuse glioma [28].
Considering the intimate relationship between GBMs and hypoxia, deep insight into genetic alterations of GBMs under hypoxic conditions is essential. Evidence has revealed that there is an intimate interplay between IDH1/2 and HIFs. Mutated IDH1/2 leads to elevated expression of oncometabolite R-2-hydroxyglutarate (2HG), which then decreases HIF1α and HIF2α levels [29]. Interestingly, IDH mutation alone inhibits tumor growth while the combinatory effect of IDH1/2 and HIF promotes neoplastic growth, contributing to unfavorable prognosis in GBM patients. It is reported that expression of monocarboxylate transporter-4 (MCT4), protein phosphatase 2A (PP2A), Krupple-like 4 (Klf4), and ATP-binding cassette B1 (ABCB1) are upregulated under hypoxic conditions and lead to shorter survival spans of GBM patients [30][31][32][33][30,31,32,33]. It is acknowledged that HIF1α level increases after mammalian target of rapamycin (mTOR) is dysregulated [34]. Multiple genes participate in the above pathways, such as PTEN (phosphatase and tensin homolog), PML (promyelocytic leukemia), and EGFR [35][36][35,36].
4. GSC and Hypoxia-Related Pathways
Accumulated research showed that numerous signaling pathways are altered in GBM, such as PI3K/AKT/mTOR, MAPK, STAT3/bcl2, PI3K/RhoA/C, HIF/IDH1/2, VEGF, EGF, Wnt/βcatenin, and Notch [37][38][39][40][37,38,39,40] (Table 1). It has been indicated that ING5 (a member of the epigenetic regulators ING family) could increase the activity of PI3K/AKT via facilitating transcription of the calcium channel as well as the follicle stimulating hormone signaling gene, to maintain self-renewal of GSCs which partially causes resistance and recurrence of GBMs [22].
Under hypoxia, there are several alterations in GBM-related pathways. Grassi et al. reported that hypoxia could induce upregulation of Delta like non-canonical Notch ligand 1 (DLK1) in GBM, thus promoting colony formation of GBMs as well as gene expression of GSC markers [41]. A recent study showed that CBF1, a cardinal transcriptional modulator of Notch signaling pathway, could be activated by hypoxia to promote proliferation of GSCs and accelerate epithelial-to-mesenchyme transition (EMT), further enhancing chemoresistance of GBMs [42].
Vascular endothelial growth factor (VEGF) is a pro-angiogenic factor that mediates vascular permeability and angioedema. VEGFR2 is a receptor of VEGF. In hypoxia, both VEGF and VEGFR2 are up-regulated by HIF1α and overexpressed in GBM, accelerating tumor progression and invasion [43]. In addition, enhanced HIF1α expression could activate the JAK1//2-STAT3 pathway that closely associates with VEGF secretion, thus promoting self-renewal of GSCs [44]. Bevacizumab is a kind of anti-VEGF monoclonal antibody currently used as a second-line agent which shows efficiency in decreasing aberrant vascularization of GBM [45]. Brefeldin A is another inhibitor of VEGF in GBM [46]. However, anti-VEGF treatments inevitably lead to therapy resistance. An investigation indicated that resistance to anti-VEGF therapy in GBM is facilitated by elevation of regulatory T-cell (Treg), which might serve as potential targets with both immunologic and anti-VEGF effects [47].
The Wnt pathway has an intimate association with GSC features, being able to reduce CD133 and Nestin under both aerobic and anaerobic conditions. In hypoxia, HIF1α upregulates expression of both TCF-1 and LEF-1 to cooperate with Wnt signaling in GBM, reprograming GSC phenotype towards a more differentiated and less aggressive one. A study has revealed that hypoxia-induced Wnt activation could inhibit Notch pathway in primary GBM and enhance chemosensitivity of GBM cells towards temozolomide (TMZ) therapy [48].
Transforming growth factor β (TGFβ) is a downstream gene of HIF1α with two isoforms TGFβ1/β2. TGFβ plays significant roles in GBM progression and recurrence [49], and can promote GSC invasion in vitro [50]. Integrin avβ8 is overexpressed in GSC and crucial for GSC self-renewal and GBM tumorigenesis. It was demonstrated that avβ8 integrin mediates GBM progression via promoting TGFβ1-induced DNA replication, thus the avβ8-integrin-TGFβ1 axis might function as a therapeutic target of GBM [51]. Dedifferentiation of non-stem cells into stem cells is known to be involved in EMT. Under hypoxia, GSCs could release TGFβ1 to promote EMT, leading to increased quantities of GSC and poor outcomes of GBM patients [49]. Interestingly, TGFβ is recognized as an upstream regulator of VEGF, and modulating VEGF and TGFβ signaling pathways collectively could effectively control neoplastic growth of GBMs [52].
Long noncoding RNA (lncRNA) H19 displays a tumorigenic role in GBMs under hypoxia. The research has highlighted that targeting lncRNA H19 might be a potential therapeutic strategy for GBMs [53]. Hypoxia-inducible and lipid droplet associated protein (HILPDA, also identified as HIG2) is inherently overexpressed in GBMs and enhanced by hypoxia, contributing to unfavorable prognosis of GBM patients [54].
5. GSC and Hypoxia-Related Metabolism
The “Warburg effect” refers to elevated levels of aerobic glycolysis in which pyruvate is transformed into lactate instead of entering Krebs cycle. Hypoxia could affect one-carbon metabolism of GSC via over-expression of aerobic glycolytic pathway enzymes such as LDHA (lactate dehydrogenase A), PFK1 (phosphofructokinase 1), and HK2, as well as down-regulation of vitamin B12 transporter protein TCN2 (Figure 1). TCN2 is indispensable in the process of GSC transformation into the highly malignant mesenchymal/CSC profile [55]. The IDH3α/cSHMT (cytosolic serine hydroxymethyltransferase) signaling axis is recognized as a novel regulatory target of one-carbon metabolism in GBM [56]. Epigenetic regulation via histone alteration, DNA methylation, and non-coding RNA could also mediate glycolytic metabolism in GBM [57]. Apart from glucose metabolism of GSCs, HIFs also play a vital role in the metabolism of amino acid. LAT1 is a transporter of branched-chain amino acid (BCAA) while BCAT1 is a metabolic enzyme of BCAA. It was reported that HIF1α and HIF2α increase both mRNA and protein levels of LAT1 and BCAT1 in GBM under hypoxia [15]. In hypoxic condition, the major carbon fuel of GBM cells partially convert from glucose to glutamine [58]. α-ketoglutarate (αKG) is a medium metabolite in tricarboxylic acid (TCA) cycle. αKG and the associated amino acid glutamate are two key factors of GBM metabolic alterations [59]. It has been identified that Acyl-CoA-Binding protein could facilitate tumorigenesis of GBMs via promoting fatty acid oxidation [60]. In conclusion, hypoxia-related metabolism is essential for the initiation and progression of GBM, which is involved in complicated processes and deserves further investigation to obtain more insights of the GSC properties.
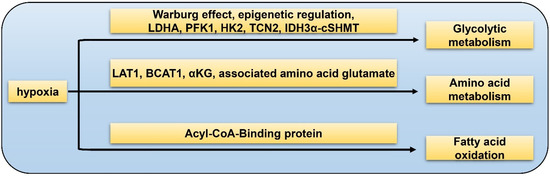
Figure 1. Hypoxia-related metabolism of GSC. Glycolytic metabolism: hypoxia promotes GSC glycolysis via Warburg effect, epigenetic regulation, LDHA, PFK1, HK2, TCN2, and IDH3α-cSHMT pathway; amino acid metabolism: hypoxia facilitates GSC glutamine metabolism by LAT1, BCAT1, αKG, and associated amino acid glutamate; fatty acid oxidation: hypoxia accelerates fatty acid metabolism through the Acyl-CoA-Binding protein.
6. GSC and Hypoxia-Related Vasculature
Vasculature plays a crucial role in GBM initiation and progression. There exist five potential mechanisms of GBM vascularization: angiogenesis, vasculogenesis, vessel mimicry, vessel co-option, and intussusception [61] (Figure 2). Angiogenesis in GBM could be attributed, to a large extent, to interplay between GSCs and endothelial cells via VEGFR, Notch, DLL-4, and nitric oxide (NO) signals. Recruitment of the bone marrow-originated endothelial progenitor cell (EPC) by SDF-1/CXCR-4 is the precondition of vasculogenesis [62]. During vascular mimicry, tumor cells constitute vascular channels which have no endothelial cells but are still able to transport erythrocytes. Proliferation of GBM was initiated by vessel co-option, followed by angiogenesis once tumor mass grew to a certain volume [63]. Intussusception is a type of vascular remodeling where a blood vessel divides into two. In vasculatures of GBMs, GSCs interact closely with adjacent cells. ECs release NO that diffuses into GSCs. GSCs generate pro-angiogenic VEGF-A to facilitate growth of ECs [64]. It has been testified that the PAX6/DLX5-WNT5A pathway might be an underlying regulator of interaction between GSC and EC in GBM [65]. Interplay between GSC and EC is important for GBM progression, which is affected by hypoxia. In spite of obvious vascularization, the microenvironment of a GBM is usually hypoxic which might be attributed to tortuous, poorly-organized, and insufficiently-perfused tumor vessels. Hypoxia increased expression of extracellular adenosine that activates the A3 adenosine receptor (A3 AR) to promote trans-differentiation of GSC into EC [66]. Hypoxia also induces fusion of GSC with EC through pseudo-endothelialization. Astrocytes mainly produce extracellular matrix (ECM) proteins such as proteoglycan, collagen, and laminin [67] (Figure 3). As important components of ECM, integrin α6, the receptor for laminin, is enriched in GSC, and promotes interactions between GSC and EC [68][69][68,69].
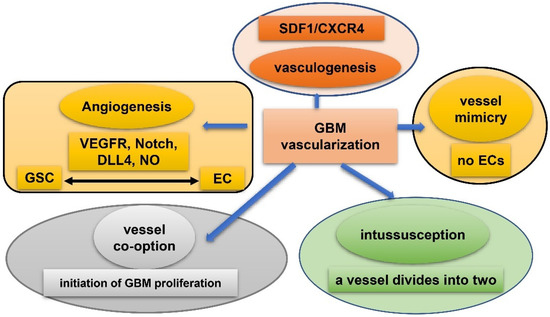
Figure 2. Five mechanisms of vascularization in glioblastoma. Angiogenesis: mainly attributed to interplay between GSCs and endothelial cells via VEGFR, Notch, DLL-4, and NO signals; vasculogenesis: bone marrow-derived endothelial progenitor cell (EPC) is recruited via SDF-1/CXCR-4 axis; vascular mimicry: tumor cells constitute vascular channels which have no endothelial cells but are still capable of transporting erythrocytes; vessel co-option: initiates proliferation of GBMs and is followed by angiogenesis; Intussusception: a blood vessel divides into two.
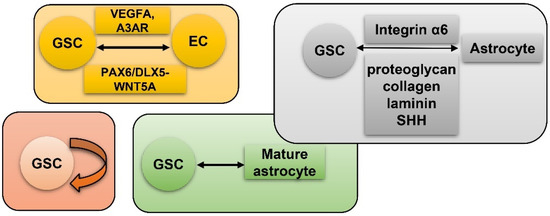
Figure 3. GSC and hypoxia-related vascularization. GSCs generate VEGFA and A3 AR to facilitate proliferation of ECs. Meanwhile, GSC and EC interact via the PAX6/DLX5-WNT5A axis; astrocytes generate proteoglycan, collagen, laminin, and SHH to interact with GSC-produced Integrin α6; self-renewal of GSC; mature GBM cell could de-differentiate into GSC.
7. GSC and Hypoxia-Related Niches
Conventional therapies mainly target CSCs, but to some extent, surrounding niches also contribute to the malignancy of neoplasm [70]. The tumor microenvironment (TME) of GBM has attracted more attention in recent years, which generally includes dendritic cells, fibroblasts, vessels, macrophages, and cancer-draining lymph nodes. TME might assist self-renewal and stemness of GSCs, acting as novel therapeutic targets. Five niches are recognized and elucidated in detail: peri-vascular niche, immune niche, hypoxia/necrotic niche, ECM niche, and peri-arteriolar niche [71]. The TME may contain more cell types that have impacts on GSCs. For example, astrocytes express Sonic hedgehog (SHH) and modulate the self-renewal of GSC and progression of GBM [72].
8. GSC and Hypoxia-Related Autophagy
Another remarkable hypoxia induced response of GBM to obtain therapy resistance and tumorigenicity is autophagy [73][74][87,108]. Autophagy is a highly-conserved catabolic reaction during evolution, which is a downstream event of mTOR hyper-activation. When oxygen is sufficient, degradation of HIF1α lead to activation of mammalian target of rapamycin (mTOR) and inhibition of autophagy. Conversely, under hypoxia, autophagy is induced through abnormal activation of Notch, Wnt/β-catenin, Hedgehog signaling pathways, and autophagy-related 9 A (ATG9A) in GBM [75][76][109,110] (Figure 47). Autophagy modulates protein degradation and turnover of neuronal stem cells (NSCs). Upregulation of autophagy could promote self-renewal and expansion of GSCs. In hypoxic condition, autophagy is also closely related to dysregulated metabolism pathways in GSCs in that autophagy provides a source of energy for tumor cells [75][109]. Autophagy functions as a protective mechanism against chemotherapy in GBM. For instance, temozolomide (TMZ) resistance in GBM is partly attributed to induced autophagy. Fortunately, scientists have identified potential novel drugs targeting autophagy. Inhibition of autophagy enhances chemosensitivity of GSCs to TMZ by igniting ferroptosis [77][111]. Tocilizumab, an inhibitor of IL6 receptor, decreases autophagy and upregulates chemosensitivity of TMZ in GBM [78][112]. GBM patients treated with chloroquine (CQ), a kind of autophagy flux suppressant, display reduced chemoresistance and better survival [79][113]. Inhibitor of the MST4-ATG4B signaling axis suppresses autophagy, which then decreases the malignancy of GBM [80][114]. Taken together, autophagy increases hypoxia-induced chemoresistance of GBM while inhibitors of autophagy have the capacity to reverse this phenomenon, being a potential therapeutic target for GBM.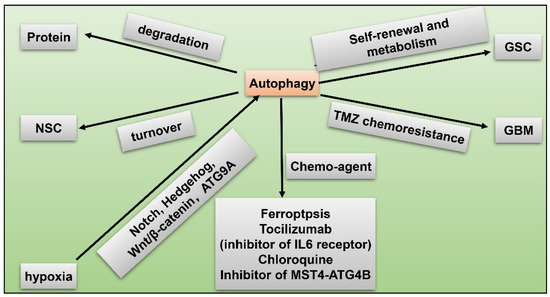
Figure 47. GSC and hypoxia-related autophagy. Hypoxia induces autophagy via activated Notch, Hedgehog, Wnt/β-catenin pathways, and autophagy-related 9 A (ATG9A) in GBM; autophagy modulates protein degradation; autophagy regulates turnover of NSCs; upregulated autophagy promotes self-renewal and metabolism of GSC; autophagy facilitates TMZ resistance in GBM; chemo-agents such as ferroptosis, tocilizumab, chloroquine, and inhibitor of MST4-ATG4B axis suppress autophagy and reduce malignancy of GBM.