Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Naser Alsharairi and Version 2 by Camila Xu.
Neonatal diabetes (NDM) is rare and presents in infants up to 6 months of age. Gestational diabetes mellitus (GDM), characterized by increased peripheral insulin resistance (IR), may cause fetal complications, including weight gain, glucose intolerance, and death. However, its effect on NDM remains uncertain.
- neonatal diabetes
- gestational diabetes
- gut microbiota
- diet
- gene expression
1. Introduction
Diabetes development in the first months of life is unlikely to be related to Type 1 diabetes (T1D) autoimmunity [1][2][1,2]. Neonatal diabetes (NDM), also termed congenital diabetes, is rare and presents in infants up to 6 months of age. It has more than 20 different monogenic causes, for example, mutations in signal transducer and activator of transcription 3 (STAT3) and LPS-responsive beige-like anchor (LRBA) genes [3][4][3,4]. NDM has been diagnosed in extremely preterm infants [5][6][7][8][5,6,7,8], particularly those with mutations in chromosome 6q24, GATA binding protein 6 (GATA6), and potassium inwardly rectifying channel, subfamily J, member 11 (KCNJ11) [7][8][7,8]. Infants diagnosed with NDM with no mutation in the KCNJ11 gene were less likely to have T1D-associated high-risk human leukocyte antigen (HLA) genotypes (DR3-DQ2/X, DR4-DQ8/X, and DR4-DQ8/DR3-DQ2) [1]. Gestational diabetes mellitus (GDM), characterized by increased peripheral insulin resistance (IR), may cause fetal complications, including weight gain, glucose intolerance, and death. However, its effect on NDM remains uncertain [9].
The colonization of the gut with microbes starts prenatally, in utero, after birth, and during breastfeeding. According to the sterile womb hypothesis, microbes are transported from the mother through the lymphatic system or blood stream into the placenta and then translocated to the fetal gut [10][11][12][10,11,12]. The maternal gut microbiota during pregnancy is crucial in shaping the composition of the gut microbiota and immune functions early in life, where diet and other factors (e.g., gestational age, maternal obesity, antibiotic usage) are found to influence the infant gut microbial diversity and richness, which in turn may enhance gut dysbiosis and disease susceptibility later in life [12][13][14][15][16][12,13,14,15,16]. Gut microbial dysbiosis in early life plays a significant role in the development of inflammation-related diseases, such as obesity, asthma, inflammatory bowel disease, and necrotizing enterocolitis [17][18][19][20][17,18,19,20].
The gut microbiota dysbiosis involved in NDM remains unclear. Compelling experimental studies reported that GDM could result in maternal gut and/or neonatal meconium microbiota dysbiosis, characterized by an increased abundance of Actinobacteria and Proteobacteria, Streptococcus, Bacteroides, Lachnospiraceae, Clostridium, Klebsiella, Desulfovibrio, Rothia, Shigella, Escherichia, Collinsella, and Proteus [21][22][23][24][25][21,22,23,24,25]. Diet has been shown to induce gut dysbiosis in women with GDM, along with gut dysbiosis in their newborns [26][27][26,27]. However, the exact mechanisms by which diet in GDM alters gut microbiota composition, which may in turn influence gene expression in NDM, are not well understood. A potential hypothesis is that epigenetic mechanisms mediate the effects of diet-microbiota interactions on altering gene expression in NDM. In general, there is a complex interaction between epigenetics, diet, and gut microbiota that can influence gene expression profiles in NDM.
2. An Overview of NDM
NDM is classified into transient (TNDM), permanent (PNDM), or syndrome types, which have expressed significant genetic changes causing persistent hyperglycemia, reduced β-cell mass or replication, delayed pancreatic islet development, and impaired insulin secretion [28]. Autosomal recessive or dominant mutations in the preproinsulin (INS) and the ATP-sensitive potassium (KATP) channel (very common to KCNJ11 or ATP-binding cassette transporter subfamily C member 8, ABCC8) are the major genes responsible for TNDM and PNDM. PNDM can also result from autosomal recessive mutations in the glucokinase (GCK) and pancreatic and duodenal homeobox l (PDX1) genes. Compound heterozygous or homozygous mutations in GCK and PDX1 may cause β-cell glucose sensing impairment and hypoplasia or pancreas agenesis. Insulin or sulfonylurea therapy can often be used for infants with mutations in KCNJ11, ABCC8, and INS [29][30][31][32][33][34][35][29,30,31,32,33,34,35]. A previous study identified a novel imprinted gene (protein phosphatase 1 regulatory subunit 13 like, PPP1R13L) on chromosome 19q13.32 that is hypomethylated in TNDM and associated with the zinc finger protein 57 homolog (ZFP57) [36]. TNDM is seen most often in cases of hyperglycemia and intrauterine growth retardation and may cause IR later in life [37]. An experimental study demonstrated that the CC dinucleotide sequence of the human INS gene’s active chromatin during pancreas development is mutated in TNDM. The CC dinucleotide mutation also results in disrupted GLI-similar 3 (GLIS3)-dependent activation of an episomal INS gene [38]. The most common cause of syndrome NDM is autosomal recessive mutations in eukaryotic translation initiation factor 2-α kinase 3 (EIF2Ak3) (diabetes associated with renal dysfunction/epiphyseal dysplasia), Solute carrier family 2 member 2 (SLC2A2) (diabetes associated with facilitated glucose transporter), Solute carrier family 19, member 2 (SLC19A2) (diabetes associated with megaloblastic anemia syndrome), Insulin receptor (INSR) (diabetes associated with severe IR), and Forkhead box P3 (FOXP3) (diabetes associated with polyendocrinopathy/immunodysregulation) [29][30][31][34][35][29,30,31,34,35]. NDM causes insulin deficiency as a result of β-cell destruction or the impaired function of β-cells [2]. Monogenic NDM is associated with growth restriction in utero because of insulin insufficiency that relies on gene mutations in the brain, which may lead to neurodevelopmental disability [39]. There is recent evidence for severe insulin deficiency, increased islet β-cell destruction, and low birthweight and C-peptide levels in infants diagnosed with a high polygenic risk in the first six months of life [40].3. Epigenetic Modifications and NDM Gene Expression Profiles in Neonates Exposed to GDM
Epigenetic changes in several genes involved in GDM are thought to impact newborn metabolic disease susceptibility [41][42][43][41,42,43]. A recent genome-wide methylation analysis identified many enriched pathways for hypo/hyper-differential methylation genes (DMGs) in the placenta and/or the umbilical cord blood of newborns exposed to maternal GDM. The top-ranking pathway enriched in 84 DMGs was the “insulin secretion/IR” pathway [44]. It has been shown that alterations of DNA methylation characterized by significant hypermethylation at two cytosine-phosphate-guanine dinucleotide (CpG) sites and hypomethylation at all CpG sites in adipose tissues of women with GDM and fetal cord blood cells are responsible for reduced adiponectin mRNA expression associated negatively with blood glucose and homeostatic model assessment-IR (HOMA-IR) [45]. This suggests that reduced adipose tissue adiponectin expression may be considered a pathogenic factor in GDM offspring [45]. A study analyzing the DNA methylation profile in the cord blood of newborns exposed to women with GDM has identified 200 differentially methylated loci. Some metabolic disease/T1D-related genes (interleukins 6 and 10; IL-6, IL-10) and pathways enriched by differentially methylated loci were identified. The top metabolically related signaling pathways, including mitogen-activated protein kinase (MAPK), Janus kinase (JAK), phosphatidylinositol-3 kinase (PI3K), and STAT3, were identified [46]. Epigenetics is considered a key mechanism that affects glucose metabolism genes involved in GDM, and their dysregulation leads to differential DNA methylation of the tribbles homolog 1 (TRIB1) gene and vasoactive intestinal peptide receptor (VIPR1) in the placenta and fetal cord blood [47]. VIPR1 is highly expressed in pancreatic β-cells, which may activate adenylate cyclase and insulin secretion by increasing intracellular cyclic AMP (cAMP) production, which in turn stimulates protein kinase A (PKA) and increases optimal calcium influx. Genetic deletion of VIPR1 could lead to glucose intolerance [48]. The TRIB1 gene, mapped to chromosome 8q24 [49], was found to be influenced by GDM exposure in the umbilical vein endothelial cells of newborns [50]. An experimental study showed that the TRIB1 gene was associated with pro-inflammatory gene cyclooxygenase-2 (COX-2) overexpression by the action of regulated early growth response gene-1 (EGR-1), which resulted in increased glucose levels in small for gestational age neonate-derived mesenchymal stem cells [51]. In pancreatic islet β-cells, COX-2 expression was associated with downregulation of PDX1-related NDM by increasing the IL-1β autostimulation [52]. The placental tissue of women with GDM induces pro-inflammatory gene expression (tumor necrosis factor-α, TNF-α) that dysregulates insulin signaling and reduces insulin secretion from β-cells under the condition of hyperglycemia [53]. Placental GDM is the major secretion site of growth hormones (e.g., insulin-like growth factor, IGF) that play a role in stimulating pro-inflammatory cytokine production by activation of inflammatory pathways, such as PI3K [53], which increases glucose levels in the fetal cord blood and may result in NDM. The results of previous studies suggest an association between the GDM intrauterine environment and placental DNA methylation [53][54][53,54]. Higher placental DNA methylation of the PPAR-γ coactivator-1-α (PGC1α) gene was associated with IR and insulin secretion in women with GDM [54]. High maternal glucose levels were reported to be associated with placental DNA methylation changes to the PGC1α gene on chromosome 4p15.1 in GDM, suggesting that PGC-1α disturbs placental functions, which may increase the risk of diabetes in offspring [55]. PGC1α mRNA expression in human adipocytes has been linked to IR markers. Patients with IR and visceral obesity have demonstrated reduced PGC1α mRNA expression in adipose tissue, which leads to increased adiponectin and IL-6 serum levels [56]. In one longitudinal study, maternal GDM was shown to induce high DNA methylation variations at the PGC1α gene locus, and such variations may mediate the impact of GDM on increasing fetal cord blood glucose levels [57]. PGC1α mRNA expression in both adipose and placenta tissue of GDM women has an impact on glucose and lipid homeostasis by increasing adiponectin and low density lipoprotein (LDL) cholesterol levels and decreasing tryglycerides and glucose levels [58]. Levels of PGC1α and PDX1 were reported to be reduced in placental tissue of women with GDM, which may lead to abnormal glucose metabolism in newborns [59]. Upregulating PGC1α activity in the brain and lungs of preterm infants has a significant role in activating the transcription factors implicated in mitochondrial biogenesis and increasing mitochondrial antioxidant enzymes, which in turn may reduce inflammation and oxidative stress (OS) by downregulating pro-inflammatory cytokines and chemokines [60]. Overexpression of the forkhead box O 1 (FoxO1) in pancreatic β-cells regulated by glucagon-like peptide 1 (GLP-1) stimulation results in inhibited PGC1α and its target gene, PDX1-related NDM [61]. The imprinted mesoderm-specific transcript (MEST) gene showed significant DNA methylation at five CpG sites in the cord blood of GDM newborns, which in turn led to decreased MEST methylation, thus influencing obesity and diabetes susceptibility [62]. A large-scale, genome-wide study has identified a total of 4485 hypermethylated and hypomethylated CpG sites in 2198 differentially methylated genes (e.g., MEST) enriched in the T1D pathway in the cord blood of infants born to GDM women [63]. MEST, located on chromosome 7q32, a gene belonging to a cluster of carboxypeptidase A (CPA) genes, has been implicated in postnatal and intrauterine growth restriction related to congenital Silver-Russell syndrome (SRS) [64]. Evidence suggests that paternal inherited H19/IGF2:IG-DMR deletions interfering with ZFP57 involved in NDM may result in SRS [65]. A case report has shown that maternal inheritance at chromosomes 2, 8, and 21 in the region of PLAG1 like zinc finger 1 (PLAG1)-associated NDM is responsible for SRS [66]. A pilot study has identified differentially methylated regions of POU class 2 homeobox 1 (POU2F1), paraoxonase 1 (PON1), and NF-E2 related factor 2 (NRF2) in the cord blood of newborns of GDM women [67]. POU2F1 is found on chromosome 1q24, a locus with evidence of strong linkage disequilibrium for its relationship to type 2 diabetes (T2D) [68]. Treatment of pancreatic β-cells with hydrogen peroxide (H2O2) results in enhanced POU2F1 activity as well as other inflammatory signaling pathway activation, such as c-jun N-terminal kinase (JNK) and DNA-dependent protein kinase (DNA-PK) [69], which in turn may increase IR and diabetes susceptibility. Methylation for the PON1 gene in mothers, which is localized on chromosome 7q21-22 [70], has been observed in children of mothers exposed to adverse life events, which coincides with the presence of the ZFP57 gene implicated in NDM [71]. The Q192R polymorphism of the PON1 gene was reported to increase GDM susceptibility, which could be a marker for IR [72]. High PON1 levels and PON1 lactonase activity were associated with increased OS, which causes alterations of the glycolipid metabolic profiles in infants born to GDM women [73]. Neonates demonstrate increased free PON1, decreased PON1 lactonase activity, and different PON1 distribution in the high-density lipoprotein (HDL) subclasses in cord blood. PON1 lactonase activity was observed to be lower in the large HDL group than in the small HDL group [74]. Impaired NRF2 activity was reported to increase IR and OS associated with diabetes, which can contribute to decreased antioxidant enzyme activity in pancreatic β-cells [75]. GDM contributes to fetal NRF2-mediated antioxidant signaling dysregulation in fetal endothelial cells by increasing OS, protein carbonylation, and mitochondrial reactive oxygen species (ROS) generation [76]. NRF2 has been shown to restore PDX1 levels in pancreatic β-cells by reducing OS-mediated JNK-dependent FOXO1 activation [77]. A candidate gene study showed that downregulation of PDX1 mRNA expression in the placentas of women with GDM resulted in increased blood glucose levels in fetal cord blood [59]. A recent meta-analysis of epigenome-wide association studies showed that maternal hyperglycemia during pregnancy is associated with reduced offspring DNA methylation at two CpG sites located in the thioredoxin interacting protein (TXNIP) gene [78]. Overexpression of TXNIP, also termed α-arrestin, in pancreatic β-cells increases glucose levels by binding to and suppressing the antioxidant protein thioredoxin (TXN), which may lead to impaired activity of the angiogenic cytokine vascular endothelial growth factor (VEGF), increased ROS expression, induction of apoptosis, and decreased insulin production [79][80][79,80]. Overexpression of TXNIP mRNA in the placenta increases ROS production and mitochondrial dysfunction as a result of decreasing TXN expression levels [81]. In one study, TXNIP mRNA expression was reported to increase in GDM women but not in neonates. On the other hand, TXN mRNA expression in the placenta was high. The thioredoxin (TXN)/TXNIP ratio increases in the placenta and neonatal cord blood of GDM women, concurrent with increased expression of nuclear factor-kappa B (NF-kB), as well as STAT3 and its target protein suppressor of cytokine signaling 3 (SOCS3) [82]. This indicates that TXN expression in the placenta may exert a protective role in protecting the newborn from oxidative effects. Another study has linked NDM with an aberrant activation of STAT3, which leads to pancreatic β-cell dysfunction and reduced insulin expression [83]. These findings suggest that exposure to GDM causes alterations in placental and/or fetal cord blood DNA methylation, which in turn may impact fetal glucose metabolism genes involved in NDM. Differentially expressed genes might be enriched in the PI3K, STAT3, JAK, and MAPK signaling pathways and other inflammatory genes, such as COX-2, IL-6, and IL-10. On this basis, seven genes related to NDM were identified: TRIB1, PGC1α, MEST, POU2F1, PON1, NRF2, and TXNIP. Figure 1 shows NDM-related gene-specific DNA methylation in neonates exposed to GDM.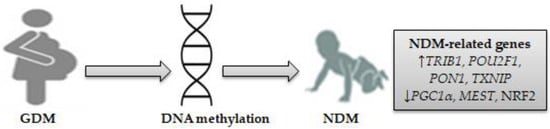
Figure 1. DNA methylation as a mechanism for NDM-related genes in neonates exposed to GDM. Seven differentially expressed genes involved in NDM were identified, consisting of four upregulated genes (TRIB1, POU2F1, PON1, TXNIP) and three downregulated genes (PGC1α, MEST, and NRF2). (↓) decrease, (↑) increase.