The family of α/β hydrolases is one of the largest known protein families, including a wide range of members such as epoxide hydrolases, dehalogenases, hydroxynitrile lyases, fungal lipases, amidases, dienelactone hydrolases, haloperoxidases, acetylcholine esterases, serine carboxypeptidases, serine carboxypeptidase-like acyltransferases and other enzymes with distinct functions. Although many natural enzymes have been screened as biocatalysts with excellent performance, most of them are still unable to meet the needs of industrial applications. Low catalytic activity, thermostability, and enantioselectivity under complex and harsh industrial process conditions are still the main limitations for the large-scale application of natural enzymes. With the development of protein engineering technology, functional improvements have been achieved for existing α/β hydrolases, specifically in key enzyme characteristics such as their enantioselectivity and stability, in order to tailor these enzymes for specific industrial applications.
- chiral compound
- α/β hydrolase
- catalytic mechanism
1. Structure and Catalytic Mechanism of α/β Hydrolase
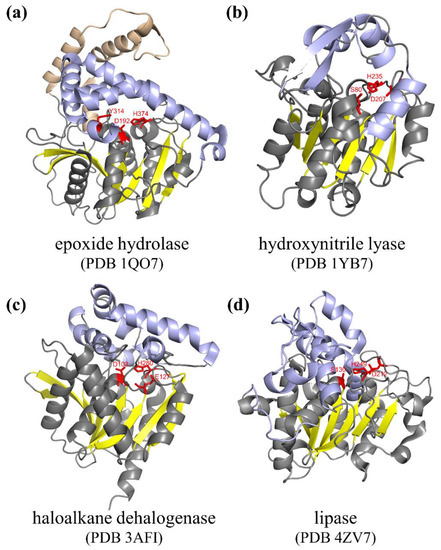
2. Engineering of Natural α/β Hydrolases
| Type | Enzyme | Source | Mutation Site | Reaction/Effects | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Epoxide hydrolase | Alp1U | Streptomyces ambofaciens | W187F/Y247F | Regioselective nucleophilic attack at C-2 of fluostatin C | [56] | [11] | |||||
| Alp1U | Streptomyces ambofaciens | Y247F | Highly regioselective attack at C-3 of fluostatin C | [56] | [11] | ||||||
| Am | EH | Agromyces mediolanus | ZJB120203 | - | Hydrolysis of ( | R | )-ECH to enantiopure ( | S | )-ECH | [49] | [12] |
| An | EH | Aspergillus niger | - | Hydrolysis of epoxides to the more water-soluble and usually less toxic diols | [22] | [7] | |||||
| An | EH | Aspergillus niger | A217L | Improvement in enantioselectivity | [57] | [13] | |||||
| An | EH | Aspergillus niger | A217V | Increase of the activity to allyl glycidyl ether | [57] | [13] | |||||
| An | EH | Aspergillus niger | A217C | Increase of the activity towards allyl glycidyl ether and styrene oxide | [57] | [13] | |||||
| Ar | EH | Agrobacterium radiobacter | AD1 | T247K/I108L/D131S | Improvement of activity, enantioselectivity, and thermostability | [58] | [14] | ||||
| Au | EH2 | Aspergillus usamii | E001 | - | Resolution of racemic styrene oxide | [47] | [15] | ||||
| Au | EH2 | Aspergillus usamii | A214C/A250I | 12.6-fold enhanced enantiomeric ratio toward | rac | -styrene oxide | [59] | [16] | |||
| Au | EH2 | Aspergillus usamii | R322V/L344C | High enantioselectivity towards | rac- | ortho-trifluoromethyl styrene oxide | [60] | [17] | |||
| EchA | Agrobacterium radiobacter | AD1 | I219F | Enhanced enantioselectivity for styrene oxide | [61] | [18] | |||||
| EchA | Agrobacterium radiobacter | AD1 | L190F | Enhanced activity for styrene oxide | [61] | [18] | |||||
| Gm | EH3 | Glycine max | - | Enantioconvergent hydrolysis of | rac | -epoxides with high enantiopurity and yield | [48] | [19] | |||
| SgcF | Streptomyces griseus | IFO 13 350 | W236Y/Q237M | 20-fold increased activity toward ( | S | )-styrene oxide to yield an ( | S | )-diol | [62] | [20] | |
| Sibe-EH | metagenomes | - | Desymmetrization of | cis | -2,3-epoxybutane producing the (2 | R | ,3 | R | )-diol | [52] | [21] |
| CH65-EH | metagenomes | - | EH activity toward a broad range of substrates and with high thermostability |
[52] | [21] | ||||||
| Vr | EH1 | Vigna radiata | - | Enantioconvergent hydrolysis of | p | -nitrostyrene oxide | [44,45] | [22][23] | |||
| Vr | EH2 | Vigna radiata | - | Enantioconvergent hydrolysis of | p | -nitrostyrene oxide | [45] | [23] | |||
| Vr | EH3 | Vigna radiata | - | High and complementary regioselectivity toward styrene oxides and high enantioselectivity toward | o | -cresyl glycidyl ether | [46] | [24] | |||
| Esterase | PFE | Pseudomonas fluorescens | replacement of a loop (A120-V139) with the corresponding element (P132-Y152) of the epoxide hydrolase EchA | Conversion of an esterase into an epoxide hydrolase towards | p | -nitrostyrene oxide | [63] | [25] | |||
| Rh | Est1 | Rhodococcus | sp. ECU1013 | circular permutation mutants with G20/T19, S22/N21, and G24&A23 as new termini, respectively | Improved thermostability | [64] | [26] | ||||
| SABP2 | Nicotiana tabacum | Q221M | Higher stability (6.6-fold half-life) | [65] | [27] | ||||||
| SABP2 | Nicotiana tabacum | G12T/M239K | Switching from an esterase to a hydroxynitrile lyase | [66] | [28] | ||||||
| Haloalkane dehalogenase | DbjA | Bradyrhizobium japonicum | USDA110 | - | Excellent enantioselectivity for α-bromoesters and high enantioselectivity for two β-bromoalkanes | [24] | [9] | ||||
| DbjA | Bradyrhizobium japonicum | USDA110 | H280F | Realized the transhalogenation reaction | [67] | [29] | |||||
| DbeA | Bradyrhizobium elkanii | USDA94 | surface loop-helix transplantation from haloalkane dehalogenase DbjA | Lower stability but increased activity with various halogenated substrates and altered its enantioselectivity | [68] | [30] | |||||
| DhaA | Rhodococcus rhodochrous | E20S/F80R/C128F/T148L/A155P/A172I/C176F/D198W/V219W/C262L/D266F | Increased thermostability (Δ | Tm | 24.6 °C) | [69] | [31] | ||||
| DhaA | Rhodococcus rhodochrous | two mutants containing 13 and 17 mutation sites, respectively | Enantioselective production of ( | R | )-and ( | S | )-2,3-dichloropropan-1-ol, respectively | [38] | [32] | ||
| LinB | Sphingomonas paucimobilis | UT26 |
E15T/A53L/A81K/F169V/A197P/D255A/A247F | Increased thermostability (Δ | Tm,app | 23 °C) | [70] | [33] | |||
| Hydroxynitrile lyase | Hb | HNL | Hevea brasiliensis | L121Y | Improved activity on an unnatural substrate mandelonitrile | [71] | [34] | ||||
| Hb | HNL | Hevea brasiliensis | T11G/E79H/K236G | Lower hydroxynitrile lyase activity and higher esterase-specific activity | [72] | [35] | |||||
| Lipase | CALB | Candida antarctica | - | Kinetic resolution of racemic alcohols and amines or desymmetrization of diols and diacetates | [25] | [10] | |||||
| CALB | Candida antarctica | a circular permutated variant of CALB with 283 /282 as the new termini | Higher catalytic activity (2.6- to 9-fold) for trans and interesterification of the different substrates | [73] | [36] |
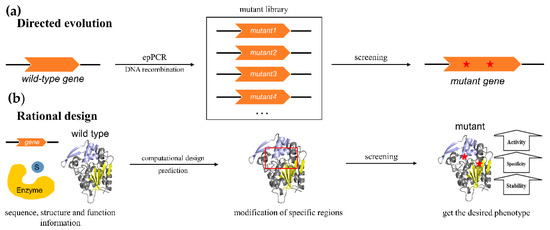
3. Engineering of α/β Hydrolases to Catalyze Different Reactions
4. Catalytic Activity Enhancement of α/β Hydrolases
5. Regio- and Stereo-Selectivity Engineering of α/β Hydrolases
6. Stability Enhancement of α/β Hydrolases
References
- Holmquist, M. Alpha/Beta-hydrolase fold enzymes: Structures, functions and mechanisms. Curr. Protein Pept. Sci. 2000, 1, 209–235.
- Lee, E.Y. Epoxide hydrolase-mediated enantioconvergent bioconversions to prepare chiral epoxides and alcohols. Biotechnol. Lett. 2008, 30, 1509–1514.
- Saini, P.; Sareen, D. An overview on the enhancement of enantioselectivity and stability of microbial epoxide hydrolases. Mol. Biotechnol. 2017, 59, 98–116.
- Schanstra, J.P.; Janssen, D.B. Kinetics of halide release of haloalkane dehalogenase: Evidence for a slow conformational change. Biochemistry 1996, 35, 5624–5632.
- Rea, D.; Fülöp, V. Prolyl oligopeptidase structure and dynamics. CNS Neurol. Disord. Drug Targets 2011, 10, 306–310.
- Pries, F.; van den Wijngaard, A.J.; Bos, R.; Pentenga, M.; Janssen, D.B. The role of spontaneous cap domain mutations in haloalkane dehalogenase specificity and evolution. J. Biol. Chem. 1994, 269, 17490–17494.
- Zou, J.; Hallberg, B.M.; Bergfors, T.; Oesch, F.; Arand, M.; Mowbray, S.L.; Jones, T.A. Structure of Aspergillus niger epoxide hydrolase at 1.8 Å resolution: Implications for the structure and function of the mammalian microsomal class of epoxide hydrolases. Structure 2000, 15, 111–122.
- Gartler, G.; Kratky, C.; Gruber, K. Structural determinants of the enantioselectivity of the hydroxynitrile lyase from Hevea brasiliensis. J. Biotechnol. 2007, 129, 87–97.
- Prokop, Z.; Sato, Y.; Brezovsky, J.; Mozga, T.; Chaloupkova, R.; Koudelakova, T.; Jerabek, P.; Stepankova, V.; Natsume, R.; van Leeuwen, J.G.; et al. Enantioselectivity of haloalkane dehalogenases and its modulation by surface loop engineering. Angew. Chem. Int. Ed. 2010, 49, 6111–6115.
- Strzelczyk, P.; Bujacz, G.D.; Kielbasinski, P.; Blaszczyk, J. Crystal and molecular structure of hexagonal form of lipase B from Candida antarctica. Acta Biochim. Pol. 2016, 63, 103–109.
- Zhang, L.; De, B.C.; Zhang, W.; Mandi, A.; Fang, Z.; Yang, C.; Zhu, Y.; Kurtan, T.; Zhang, C. Mutation of an atypical oxirane oxyanion hole improves regioselectivity of the α/β-fold epoxide hydrolase Alp1U. J. Biol. Chem. 2020, 295, 16987–16997.
- Xue, F.; Liu, Z.-Q.; Zou, S.-P.; Wan, N.-W.; Zhu, W.-Y.; Zhu, Q.; Zheng, Y.-G. A novel enantioselective epoxide hydrolase from Agromyces mediolanus ZJB120203: Cloning, characterization and application. Process. Biochem. 2014, 49, 409–417.
- Kotik, M.; Stepanek, V.; Kyslik, P.; Maresova, H. Cloning of an epoxide hydrolase-encoding gene from Aspergillus niger M200, overexpression in E. coli, and modification of activity and enantioselectivity of the enzyme by protein engineering. J. Biotechnol. 2007, 132, 8–15.
- Zou, S.P.; Zheng, Y.G.; Wu, Q.; Wang, Z.C.; Xue, Y.P.; Liu, Z.Q. Enhanced catalytic efficiency and enantioselectivity of epoxide hydrolase from Agrobacterium radiobacter AD1 by iterative saturation mutagenesis for (R)-epichlorohydrin synthesis. Appl. Microbiol. Biotechnol. 2018, 102, 733–742.
- Hu, D.; Tang, C.-D.; Yang, B.; Liu, J.-C.; Yu, T.; Deng, C.; Wu, M.-C. Expression of a novel epoxide hydrolase of Aspergillus usamii E001 in Escherichia coli and its performance in resolution of racemic styrene oxide. J. Ind. Microbiol. Biotechnol. 2015, 42, 671–680.
- Hu, D.; Hu, B.C.; Hou, X.D.; Zhang, D.; Lei, Y.Q.; Rao, Y.J.; Wu, M.C. Structure-guided regulation in the enantioselectivity of an epoxide hydrolase to produce enantiomeric monosubstituted epoxides and vicinal diols via kinetic resolution. Org. Lett. 2022, 24, 1757–1761.
- Wen, Z.; Hu, D.; Hu, B.C.; Zhang, D.; Huang, J.F.; Wu, M.C. Structure-guided improvement in the enantioselectivity of an Aspergillus usamii epoxide hydrolase for the gram-scale kinetic resolution of ortho-trifluoromethyl styrene oxide. Enzym. Microb. Technol. 2021, 146, 109778.
- Rui, L.; Cao, L.; Chen, W.; Reardon, K.F.; Wood, T.K. Protein engineering of epoxide hydrolase from Agrobacterium radiobacter AD1 for enhanced activity and enantioselective production of (R)-1-phenylethane-1,2-diol. Appl. Environ. Microbiol. 2005, 71, 3995–4003.
- Zhang, C.; Li, C.; Zhu, X.X.; Liu, Y.Y.; Zhao, J.; Wu, M.C. Highly regio- and enantio-selective hydrolysis of two racemic epoxides by GmEH3, a novel epoxide hydrolase from Glycine max. Int. J. Biol. Macromol. 2020, 164, 2795–2803.
- Horsman, G.P.; Lechner, A.; Ohnishi, Y.; Moore, B.S.; Shen, B. Predictive model for epoxide hydrolase-generated stereochemistry in the biosynthesis of nine-membered enediyne antitumor antibiotics. Biochemistry 2013, 52, 5217–5224.
- Ferrandi, E.E.; Sayer, C.; De Rose, S.A.; Guazzelli, E.; Marchesi, C.; Saneei, V.; Isupov, M.N.; Littlechild, J.A.; Monti, D. New thermophilic α/β class epoxide hydrolases found in metagenomes from hot environments. Front. Bioeng. Biotechnol. 2018, 6, 144.
- Zhu, Q.Q.; He, W.H.; Kong, X.D.; Fan, L.Q.; Zhao, J.; Li, S.X.; Xu, J.H. Heterologous overexpression of Vigna radiata epoxide hydrolase in Escherichia coli and its catalytic performance in enantioconvergent hydrolysis of p-nitrostyrene oxide into (R)-p-nitrophenyl glycol. Appl. Microbiol. Biotechnol. 2014, 98, 207–218.
- Xu, W.; Xu, J.-H.; Pan, J.; Gu, Q.; Wu, X.-Y. Enantioconvergent hydrolysis of styrene epoxides by newly discovered epoxide hydrolases in mung bean. Org. Lett. 2006, 8, 1737–1740.
- Hu, D.; Tang, C.; Li, C.; Kan, T.; Shi, X.; Feng, L.; Wu, M. Stereoselective hydrolysis of epoxides by reVrEH3, a novel Vigna radiata epoxide hydrolase with high enantioselectivity or high and complementary regioselectivity. J. Agric. Food Chem. 2017, 65, 9861–9870.
- Jochens, H.; Stiba, K.; Savile, C.; Fujii, R.; Yu, J.G.; Gerassenkov, T.; Kazlauskas, R.J.; Bornscheuer, U.T. Converting an esterase into an epoxide hydrolase. Angew. Chem. Int. Ed. 2009, 48, 3532–3535.
- Li, F.; Luan, Z.; Chen, Q.; Xu, J.; Yu, H. Rational selection of circular permutation sites in characteristic regions of the α/β-hydrolase fold enzyme RhEst1. J. Mol. Catal. B: Enzym. 2016, 125, 75–80.
- Jones, B.J.; Lim, H.Y.; Huang, J.; Kazlauskas, R.J. Comparison of five protein engineering strategies for stabilizing an α/β-hydrolase. Biochemistry 2017, 56, 6521–6532.
- Padhi, S.K.; Fujii, R.; Legatt, G.A.; Fossum, S.L.; Berchtold, R.; Kazlauskas, R.J. Switching from an esterase to a hydroxynitrile lyase mechanism requires only two amino acid substitutions. Chem. Biol. 2010, 17, 863–871.
- Beier, A.; Damborsky, J.; Prokop, Z. Transhalogenation catalysed by haloalkane dehalogenases engineered to stop natural pathway at intermediate. Adv. Synth. Catal. 2019, 361, 2438–2442.
- Marek, M.; Chaloupkova, R.; Prudnikova, T.; Sato, Y.; Rezacova, P.; Nagata, Y.; Kuta Smatanova, I.; Damborsky, J. Structural and catalytic effects of surface loop-helix transplantation within haloalkane dehalogenase family. Comput. Struct. Biotechnol. J. 2020, 18, 1352–1362.
- Bednar, D.; Beerens, K.; Sebestova, E.; Bendl, J.; Khare, S.; Chaloupkova, R.; Prokop, Z.; Brezovsky, J.; Baker, D.; Damborsky, J. FireProt: Energy- and evolution-based computational design of thermostable multiple-point mutants. PLoS Comput. Biol. 2015, 11, e1004556.
- van Leeuwen, J.G.; Wijma, H.J.; Floor, R.J.; van der Laan, J.M.; Janssen, D.B. Directed evolution strategies for enantiocomplementary haloalkane dehalogenases: From chemical waste to enantiopure building blocks. ChemBioChem 2012, 13, 137–148.
- Floor, R.J.; Wijma, H.J.; Colpa, D.I.; Ramos-Silva, A.; Jekel, P.A.; Szymanski, W.; Feringa, B.L.; Marrink, S.J.; Janssen, D.B. Computational library design for increasing haloalkane dehalogenase stability. ChemBioChem 2014, 15, 1660–1672.
- von Langermann, J.; Nedrud, D.M.; Kazlauskas, R.J. Increasing the reaction rate of hydroxynitrile lyase from Hevea brasiliensis toward mandelonitrile by copying active site residues from an esterase that accepts aromatic esters. ChemBioChem 2014, 15, 1931–1938.
- Nedrud, D.M.; Lin, H.; Lopez, G.; Padhi, S.K.; Legatt, G.A.; Kaz-Lauskas, R.J. Uncovering divergent evolution of α/β-hydrolases: A surprising residue substitution needed to convert Hevea brasiliensis hydroxynitrile lyase into an esterase. Chem. Sci. 2014, 5, 4265–4277.
- Yu, Y.; Lutz, S. Improved triglyceride transesterification by circular permuted Candida antarctica lipase B. Biotechnol. Bioeng. 2010, 105, 44–50.
- Paye, M.F.; Rose, H.B.; Robbins, J.M.; Yunda, D.A.; Cho, S.; Bommarius, A.S. A high-throughput pH-based colorimetric assay: Application focus on alpha/beta hydrolases. Anal. Biochem. 2018, 549, 80–90.
- Zheng, Y.C.; Ding, L.Y.; Jia, Q.; Lin, Z.; Hong, R.; Yu, H.L.; Xu, J.H. A high-throughput screening method for the directed evolution of hydroxynitrile lyase towards cyanohydrin synthesis. ChemBioChem 2021, 22, 996–1000.
- Glasner, M.E.; Truong, D.P.; Morse, B.C. How enzyme promiscuity and horizontal gene transfer contribute to metabolic innovation. FEBS J. 2020, 287, 1323–1342.
- Kapoor, M.; Gupta, M.N. Lipase promiscuity and its biochemical applications. Process. Biochem. 2012, 47, 555–569.
- Cadet, F.; Fontaine, N.; Li, G.; Sanchis, J.; Ng Fuk Chong, M.; Pandjaitan, R.; Vetrivel, I.; Offmann, B.; Reetz, M.T. A machine learning approach for reliable prediction of amino acid interactions and its application in the directed evolution of enantioselective enzymes. Sci. Rep. 2018, 8, 16757.
- Bommarius, A.S.; Paye, M.F. Stabilizing biocatalysts. Chem. Soc. Rev. 2013, 42, 6534–6565.