Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Eftekhar Eftekharpour.
Tau, a member of the microtubule-associated proteins, is a known component of the neuronal cytoskeleton; however, in the brain tissue, it is involved in other vital functions beyond maintaining the cellular architecture. The pathologic tau forms aggregates inside the neurons and ultimately forms the neurofibrillary tangles. Intracellular and extracellular accumulation of different tau isoforms, including dimers, oligomers, paired helical filaments and tangles, lead to a highly heterogenous group of diseases named “Tauopathies”.
- tau
- tauopathy
- neurodegeneration
- Alzheimer’s disease
- dementia
- aging
- MAPT
- cytoskeleton
1. Tauopathies: Types and Importance
Tauopathies are a group of progressive neurodegenerative diseases characterized by tau protein inclusions in the human brain. Clinically, there are twenty-six different types of tauopathies that are recognized by complicated neurodegenerative symptoms, including dementia. A series of cognitive/behavioral and memory deficits manifest in individuals affected by tauopathies [1]. From a clinical pathology perspective, those neurodegenerative diseases in which tau protein plays the main pathophysiological role are classified as “primary tauopathies”. Pick’s disease (PiD), progressive supranuclear palsy (PSP), argyrophilic grain disease (AGD), primary age-related tauopathy (PART), neurofibrillary tangle dementia (NTD-dementia) and corticobasal degeneration (CBD) are several examples of primary tauopathies. On the other hand, for the “secondary tauopathies” tau protein aggregation is not the major neurodegenerative mechanism. For instance, Alzheimer’s disease (AD), the most common form of dementia, is known as a secondary tauopathy in which tau aggregation co-exists with accumulation of β-amyloid peptide (Aβ) [2]. Chronic traumatic encephalopathy (CTE) is another form of secondary tauopathy in which tau depositions are present in neurons, astrocytes and neurites around the blood vessels but are associated with other pathophysiological features [3].
According to the World Health Organization, nearly 50 million people are currently suffering from dementia, and each year, around 10 million new cases are diagnosed with dementia. It is predicted that by 2050, the number of patients will approximately reach 140 million worldwide. Due to the debilitating nature of dementia, these patients require constant care, which significantly impacts their careers, their families and society. In 2017, WHO proposed a “Global action plan on the public health response to dementia 2017–2025” in which dementia has been identified as a public health priority [4]. This rationalizes the need for better understanding all aspects of dementia’s pathophysiology, including tauopathies.
2. Tau
Tau protein, a member of the microtubule-associated protein family, is encoded by the microtubule-associated protein tau gene (MAPT) (Gene ID: 4137), located on human chromosome 17q21.31. Tau is primarily responsible for maintaining microtubules’ stability and promoting their assembly in axons. In the central nervous system (CNS), tau is mostly found in cortical and hippocampal neurons and, to a lesser degree, in astrocytes and oligodendrocytes [5]. Tau is not specific to the CNS, but it is also found in the peripheral nervous system (PNS), both intracellularly and extracellularly. The presence of tau protein has been reported in the interstitial fluid and cerebrospinal fluid (CSF) [6]. Structurally, there are four tau protein domains, including the N- and C-terminal domains, the proline-rich domain and the microtubule-binding domain. The proline-rich domain participates in the cell signaling process, as it is particularly the target of protein kinases. Phosphorylation of serin and threonine residues, which are mainly located in this domain can affect tau binding affinity in the microtubule-binding domain. The N-terminal domain may regulate the distance between microtubules. In contrast, the C-terminal domain is involved in microtubule polymerization [7].
The MAPT gene contains 16 exons [8], but its complex and highly regulated splicing results in a variety of developmental stage-specific messenger RNA (mRNA) species, which are mostly present in the brain tissue. In the adult human brain, this alternative splicing generates six isoforms, ranging from 352 to 441 amino acids [9]. Alternative splicing in exons 2 and 3 leads to three different isoforms, including 0N, 1N and 2N. Two additional isoforms, 3R and 4R, are also generated when splicing occurs in exon 10, resulting in the production of two proteins with three and four microtubule-binding domains, respectively. The impact of MAPT alternative splicing on human brain development and pathology has not been adequately investigated and may prove to be important.
Expression levels of various tau isoforms reflect neuronal maturation state and axonal growth capability. Lower levels of tau expression have been reported in immature regions of the human brain, such as the ganglionic eminence and the rhombic lip, while tau expression pattern and splicing are slightly different in more mature brain regions [10]. Additionally, in the human prenatal brain, an abrupt shift happens in MAPT exons 2 and 10 expressions. This shift is evolutionary conserved and can be a crucial step in the transition to mature neurons. Exon 3 undergoes small temporal variations compared to the exons 2 and 10 [10]. Specifically, during early embryonic stages, the predominant tau isoform is 0N3R. Healthy adult human brains contain a balanced level of 4R and 3R isoforms. The other expressing human tau isoforms include 1N3R, 2N3R, 0N4R, 1N4R and 2N4R; the latter is the full-length form of tau protein. The 2N4R isoform contains both inserts of exons 2 and 3 in the N-terminal domain and all four microtubule-binding regions in the C-terminal domain. Collectively, this longest tau isoform includes all 441 amino acids [11], (Figure 1).
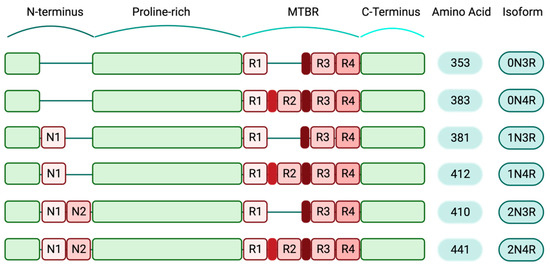
Figure 1. Human-specific tau isoforms (created with BioRender.com). Schematic diagram depicting the composition of different tau isoforms. The shortest tau variant, which is mainly expressed during the human embryonic stage, consists of 352 amino acids (0N3R) and the full-length isoform (2N4R) includes all structural domains of tau protein and 441 amino acids. Structurally, tau isoforms are comprised of N-terminus regions that contain two inserts, N1 and N2. The au N-terminals are followed by the proline-rich domains, the microtubule-binding regions (MTBR) and the C-terminals. In the full-length tau isoform, all four repeat domains (R1–4) are present in the MTBRs.
3. Physiologic Tau
Tau is an important component of the neuronal cytoskeletal compartment. The cellular skeleton is mainly composed of microtubules and is vital for the regulation of critical cell processes, including maintaining the cell shape, proper cell division, and healthy intracellular transport of organelles. In neurons, microtubular organization is critical for axonal stability and the trafficking of materials and organelles to and from the cell body. The dynamic of the cytoskeleton is highly dependent on microtubule-associated proteins (MAPs) [12], and amongst these, tau protein plays a critical role. Weingarten et al., in their milestone discovery, were able to identify tau protein as an essential factor for microtubule polymerization and microtubule assembly and dynamics [13]. As the microtubule dynamic network is essential for neurite formation and axonal pathfinding, the importance of tau protein has also been reported during neuronal development and CNS maintenance [14,15][14][15]. While tau protein expression is sharply increased during embryonic stages, this seems to reach a plateau in the mature rodent brain [16]. Authors showed that post-translational modification (PTM) of tau and its site-specific phosphorylation were differentially regulated between developing and mature brains [16]. While some residues were more phosphorylated in mature brain, overall, there was a decrease in tau phosphorylation in mature brain that might be due to an increase in tau-specific phosphatases [16].
The microtubule-binding inserts in the C-terminal domain of tau protein are the critical regions not only for binding to the microtubules but also for interacting with the actin filaments in the cytoskeleton structure [17,18][17][18]. Actin bundling is regulated by short fragments of the microtubule-binding domain in the C-terminal region of tau protein. In cell-free experiments, a synthetic version of the C-terminal region was able to bind to both monomeric and filamentous actin, but this interaction did not result in actin bundling. The authors showed that MAP2 and tau are capable of bundling actin [19]. The tau/f-actin interaction is also an important player in dendritic morphology and post-synaptic structure, therefore directly affecting synaptic stability [20]. Tau PTMs such as acetylation can modulate actin polymerization and cause synaptic dysfunction [21]. Formation of f-actin bundles is mainly produced by high-affinity interaction between proline-rich and microtubule-binding domains on tau protein and the filamentous actin. This is mediated by hydrophobic interactions between the tau microtubule-binding regions and actin filaments. On the other hand, an electrostatic bond also exists between tau proline-rich domain and f-actin. These offer a highly dynamic and multivalent interaction between tau and f-actin complexes, as the tau residues serve as “flexible linkers”. Structurally, one tau molecule contains seven actin-binding regions and these “flexible linkers” are located between these actin-binding sites [22]. Conformational changes in the actin-binding segments have a regulatory role for cross-linking between the three components of the cytoskeleton: tau, actin filaments and microtubules. Many aspects of the tau/f-actin complex, such as the exact binding sites, the bundling mechanism and the cross-linking between actin, tau and microtubules, are yet to be understood [22].
Neurons are structurally, morphologically and functionally distinct from any other cells. They have developed long processes and maintain an intricate functional connection through synapses with other proximal and distal target cells. This has evolutionarily forced them to have a sophisticated trafficking system for the transport of nutrients, energy and synaptic vesicles. “Fast axonal transport” is a complex intracellular trafficking process by which neurons transport the synthesized proteins in the somatodendritic compartment to the synapses or haul away the recycled materials and organelles. Axonal microtubules, through their molecular motors, kinesins and dyneins, play a crucial role in this bidirectional transport. While kinesin is used for transporting the cargo towards the cell periphery, dyneins are responsible for moving the cargo centrally towards the cell body, allowing for bidirectional traffic, navigating the crowded cytoplasmic environment and correctly conducting the transport tasks [23]. The connection between tau protein and microtubules can directly regulate this special ability of kinesin and dynein motors. This has been recently proposed to be mediated by the differential interaction of tau with different isoforms of dynein and kinesins. While kinesin-1 is more sensitive to inhibition by tau, kinesisn-2 and dynein are only inhibited at very high concentrations of tau. The overall sum of the connection between tau protein and microtubules regulates the forward movement or processivity of kinesin and dynein and therefore can affect the directional bias of axonal trafficking [24]. Of note, this kinesin-driven transport is tau phosphorylation-dependent, and inhibition of glycogen synthase kinase-3β (GSK3β) and reduction of tau phosphorylation is detrimental for axonal transport [25]. Increased GSK3β activity has been documented in AD, which can explain the hyperphosphorylation of tau and its detrimental effect on intracellular traffic in neurodegenerative diseases. In this active transport mode that is mediated by a tau-bound microtubule/actin network, tau is assumed to be stationary, although it is in a dynamic equilibrium with free tau molecules in the cytoplasm that are believed to be able to freely diffuse in the cytosol and axoplasm. This form of diffusion is one-directional and dependent upon the existing microtubule network. There is no need for external energy sources, and for short distances up to 1 μm, this form of tau diffusion is faster than motor-driven active transport. In this model, diffusing tau molecules occurs using the microtubule lattice to ensure proper distribution of tau molecules at different sites, i.e., somatic or axonal ends. This ensures that tau is always available for anterograde or retrograde transport systems [26].
Tau protein plays a critical role in post-synaptic scaffolding in dendrites. It interacts with the tyrosine kinase Fyn through its phosphatase activating domain (PAD), which is located on tau’s extreme N-terminal domain. The tyrosine kinase Fyn is responsible for phosphorylation of N-methyl-D-aspartate (NMDA) receptors, implying the impact of tau protein in synaptic signaling [27]. Moreover, in oligodendrocytes, tau-Fyn interaction affects the process outgrowth and is important in the initiation of axon myelination by oligodendrocytes [28].
Tau protein is not restricted to dendrites and axons, as it is also found in neuronal nuclei. In this compartment, tau serves as a DNA protection element against peroxidation through co-localizing with AT-rich heterochromatin regions of DNA and nucleoli; therefore, it contributes to the genomic stability and preservation of genomic organization [29,30,31][29][30][31].
4. Pathologic Tau
In 1906, Dr. Alois Alzheimer, a German psychiatrist and neuropathologist, described a five-year study on a clinical case with peculiar neuroanatomic features. His 50-year-old female patient suffered from paranoia, memory disturbances, sleep disorders and progressive confusion. After her death, Dr. Alzheimer investigated her brain autopsy and discovered intracellular neurofibrillary tangles (NFTs) and, consequently, described AD as an “unusual illness of the cerebral cortex” [32]. In 1963, these NFTs were characterized in the cortical neurons of the cerebrum in AD cases. These studies showed that NFTs are predominantly composed of insoluble fibers called paired helical filaments (PHFs) [33]. Two decades later, immunological assessments showed that “hyperphosphorylated tau aggregates” are the major components of PHFs [34]. Recent discoveries related to the tau protein and its pathological forms confirmed its key role in modulating neuronal physiology. To gain a better understanding of tau pathology, various aspects of this protein should be considered, including tau structure and distribution, its exact subcellular locations, possible posttranslational modifications and disease-specific isoforms.
5. One Protein and Various Conformers
Examination of tau aggregates obtained from different types of tauopathies revealed that tau filaments and inclusions are widely different in various types of tauopathies. These observations suggest that variations in tauopathy-related symptoms and also disease progression may be related to the specific pattern of tau aggregation. Tau filaments show astonishing variation in aggregation patterns, both in in vitro cell-free conditions and different tauopathy cases [35,36][35][36]. The predominant isoform of tau filaments in tau inclusions is used for the classification of different tauopathies (Figure 2) [37,38][37][38]. The complexity of different disease-specific tau structures may be a contributing factor in the complexity of tauopathies and finding treatments.
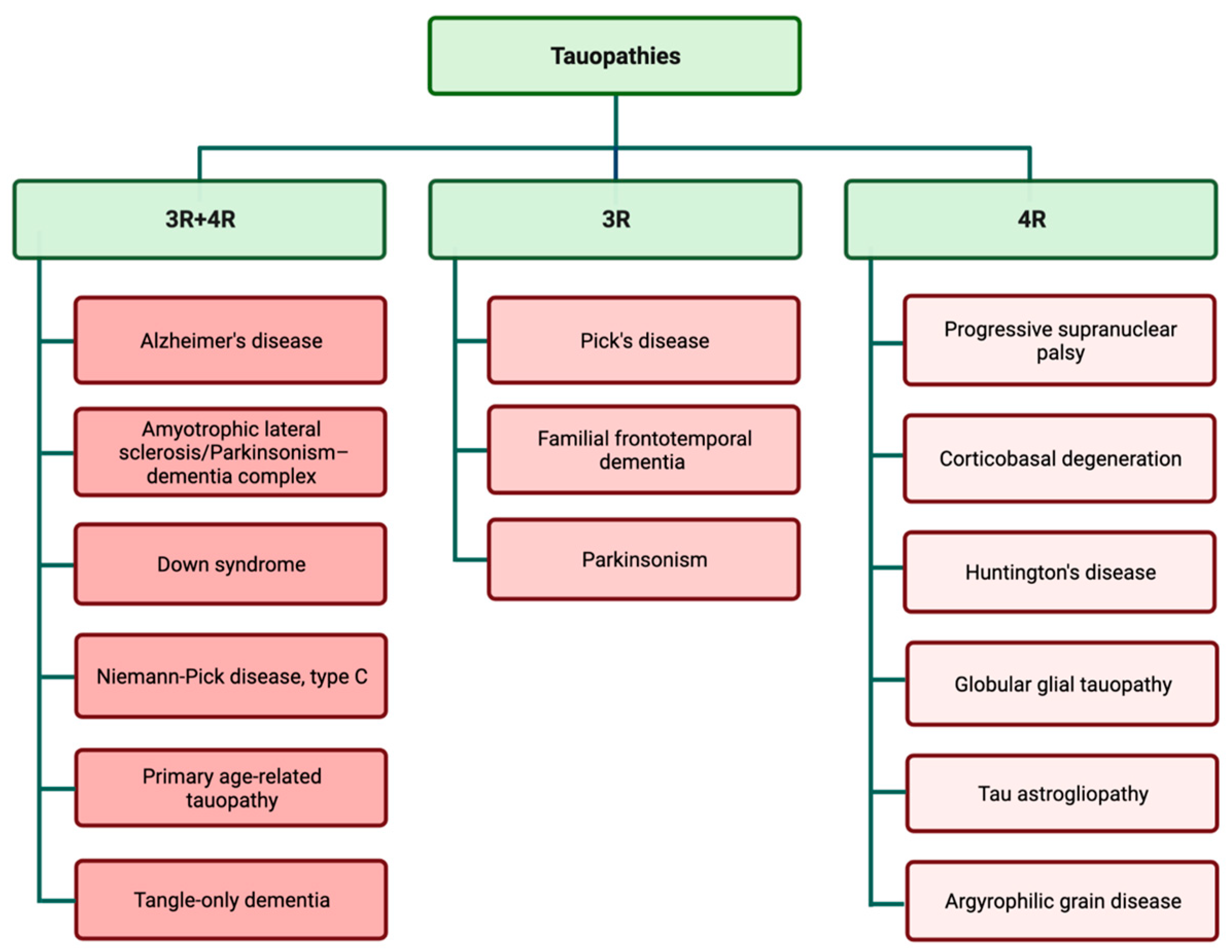
Figure 2.
Tauopathies are classified according to the presence of 3R and 4R isoforms in tau inclusions.
References
- Sexton, C.; Snyder, H.; Beher, D.; Boxer, A.L.; Brannelly, P.; Brion, J.P.; Buée, L.; Cacace, A.M.; Chételat, G.; Citron, M.; et al. Current Directions in Tau Research: Highlights from Tau 2020. Alzheimer’s Dement. 2021, 18, 988–1007.
- Gallo, D.; Ruiz, A.; Sánchez-Juan, P. Genetic Architecture of Primary Tauopathies. Neuroscience, 2022; in press.
- McKee, A.C.; Cairns, N.J.; Dickson, D.W.; Folkerth, R.D.; Dirk Keene, C.; Litvan, I.; Perl, D.P.; Stein, T.D.; Vonsattel, J.P.; Stewart, W.; et al. The First NINDS/NIBIB Consensus Meeting to Define Neuropathological Criteria for the Diagnosis of Chronic Traumatic Encephalopathy. Acta Neuropathol. 2016, 131, 75–86.
- WHO. Dementia. Available online: https://www.who.int/health-topics/dementia#tab=tab_2 (accessed on 27 August 2022).
- Sjölin, K.; Kultima, K.; Larsson, A.; Freyhult, E.; Zjukovskaja, C.; Alkass, K.; Burman, J. Distribution of Five Clinically Important Neuroglial Proteins in the Human Brain. Mol. Brain 2022, 15, 52.
- Blennow, K.; Wallin, A.; Agren, H.; Spenger, C.; Siegfried, J.; Vanmechelen, E. Tau Protein in Cerebrospinal Fluid: A Biochemical Marker for Axonal Degeneration in Alzheimer Disease? Mol. Chem. Neuropathol. 1995, 26, 231–245.
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and Tauopathies. Brain Res. Bull. 2016, 126, 238–292.
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of CDNA Clones for the Human Microtubule-Associated Protein Tau and Chromosomal Localization of the Genes for Tau and Microtubule-Associated Protein 2. Brain Res. 1986, 387, 271–280.
- Goedert, M.; Spillantini, M.; Jakes, R.; Rutherford, D. Multiple Isoforms of Human Microtubule-Associated Protein Tau: Sequences and Localization in Neurofibrillary Tangles of Alzheimer’s Disease. Neuron 1989, 3, 519–526.
- Hefti, M.M.; Farrell, K.; Kim, S.H.; Bowles, K.R.; Fowkes, M.E.; Raj, T.; Crary, J.F. High-Resolution Temporal and Regional Mapping of MAPT Expression and Splicing in Human Brain Development. PLoS ONE 2018, 13, e0195771.
- Hanger, D.; Anderton, B.; Medicine, W.N.-T. Tau Phosphorylation: The Therapeutic Challenge for Neurodegenerative Disease. Trends Mol. Biol. 2009, 15, 112–119.
- Hirokawa, N.; Takemura, R. Molecular Motors and Mechanisms of Directional Transport in Neurons. Nat. Rev. Neurosci. 2005, 6, 201–214.
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862.
- Brandt, R.; Lee, G. The Balance Between τ Protein’s Microtubule Growth and Nucleation Activities: Implications for the Formation of Axonal Microtubules. J. Neurochem. 1993, 61, 997–1005.
- Drubin, D.; Kirschner, M.W. Tau Protein Function in Living Cells. J. Cell Biol. 1986, 103, 2739–2746.
- Yu, Y.; Run, X.; Liang, Z.; Li, Y.; Liu, F.; Liu, Y.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Developmental Regulation of Tau Phosphorylation, Tau Kinases, and Tau Phosphatases. J. Neurochem. 2009, 108, 1480–1494.
- Moraga, D.M.; Nuñez, P.; Garrido, J.; Maccioni, R.B. A τ Fragment Containing a Repetitive Sequence Induces Bundling of Actin Filaments. J. Neurochem. 1993, 61, 979–986.
- Elie, A.; Prezel, E.; Guérin, C.; Denarier, E.; Ramirez-Rios, S.; Serre, L.; Andrieux, A.; Fourest-Lieuvin, A.; Blanchoin, L.; Arnal, I. Tau Co-Organizes Dynamic Microtubule and Actin Networks. Sci. Rep. 2015, 5, 9967.
- Correas, I.; Padilla, R.; Journal, J.A.-B. The Tubulin-Binding Sequence of Brain Microtubule-Associated Proteins, Tau and MAP-2, Is Also Involved in Actin Binding. Biochem. J. 1990, 269, 61–64.
- Frandemiche, M.L.; De Seranno, S.; Rush, T.; Borel, E.; Elie, A.; Arnal, I.; Lanté, F.; Buisson, A. Activity-Dependent Tau Protein Translocation to Excitatory Synapse Is Disrupted by Exposure to Amyloid-Beta Oligomers. J. Neurosci. 2014, 34, 6084–6097.
- Tracy, T.E.; Sohn, P.D.; Minami, S.S.; Wang, C.; Min, S.-W.; Li, Y.; Zhou, Y.; Le, D.; Lo, I.; Ponnusamy, R.; et al. Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron 2016, 90, 245–260.
- Cabrales Fontela, Y.; Kadavath, H.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Multivalent Cross-Linking of Actin Filaments and Microtubules through the Microtubule-Associated Protein Tau. Nat. Commun. 2017, 8, 1981.
- Orr, M.E.; Sullivan, A.; Frost, B. A Brief Overview of Tauopathy: Causes, Consequences, and Therapeutic Strategies. Trends Pharmacol. Sci. 2017, 38, 637–648.
- Chaudhary, A.R.; Berger, F.; Berger, C.L.; Hendricks, A.G. Tau Directs Intracellular Trafficking by Regulating the Forces Exerted by Kinesin and Dynein Teams. Traffic 2018, 19, 111–121.
- Cuchillo-Ibanez, I.; Seereeram, A.; Byers, H.L.; Leung, K.-Y.; Ward, M.A.; Anderton, B.H.; Hanger, D.P. Phosphorylation of Tau Regulates Its Axonal Transport by Controlling Its Binding to Kinesin. FASEB J. 2008, 22, 3186–3195.
- Hinrichs, M.H.; Jalal, A.; Brenner, B.; Mandelkow, E.; Kumar, S.; Scholz, T. Tau Protein Diffuses along the Microtubule Lattice. J. Biol. Chem. 2012, 287, 38559–38568.
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397.
- Klein, C.; Kramer, E.M.; Cardine, A.M.; Schraven, B.; Brandt, R.; Trotter, J. Process Outgrowth of Oligodendrocytes Is Promoted by Interaction of Fyn Kinase with the Cytoskeletal Protein Tau. J. Neurosci. 2002, 22, 698–707.
- Sjöberg, M.K.; Shestakova, E.; Mansuroglu, Z.; Maccioni, R.B.; Bonnefoy, E. Tau Protein Binds to Pericentromeric DNA: A Putative Role for Nuclear Tau in Nucleolar Organization. J. Cell Sci. 2006, 119, 2025–2034.
- Maina, M.B.; Bailey, L.J.; Wagih, S.; Biasetti, L.; Pollack, S.J.; Quinn, J.P.; Thorpe, J.R.; Doherty, A.J.; Serpell, L.C. The Involvement of Tau in Nucleolar Transcription and the Stress Response. Acta Neuropathol. Commun. 2018, 6, 70.
- Mansuroglu, Z.; Benhelli-Mokrani, H.; Marcato, V.; Sultan, A.; Violet, M.; Chauderlier, A.; Delattre, L.; Loyens, A.; Talahari, S.; Bégard, S.; et al. Loss of Tau Protein Affects the Structure, Transcription and Repair of Neuronal Pericentromeric Heterochromatin. Sci. Rep. 2016, 6, 33047.
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English Translation of Alzheimer’s 1907 Paper, “Uber Eine Eigenartige Erkankung Der Hirnrinde”. Clin. Anat. 1995, 8, 429–431.
- Terry, R.D. The Fine Structure of Neurofibrillary Tangles in Alzheimer’s Disease. J. Neuropathol. Exp. Neurol. 1963, 22, 629–642.
- Brion, J.P.; Passareiro, H.; Nunez, J.; Flamentdurand, J. Immunological Detection of Tau Protein in Neurofibrillary Tangles of Alzheimers-Disease. Arch. Biol. 1985, 96, 229–235.
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-Synuclein Filaments from Multiple System Atrophy. Nature 2020, 585, 464–469.
- Zhang, W.; Falcon, B.; Murzin, A.G.; Fan, J.; Crowther, R.A.; Goedert, M.; Scheres, S.H. Heparin-Induced Tau Filaments Are Polymorphic and Differ from Those in Alzheimer’s and Pick’s Diseases. Elife 2019, 8, e43584.
- Rösler, T.W.; Tayaranian Marvian, A.; Brendel, M.; Nykänen, N.P.; Höllerhage, M.; Schwarz, S.C.; Hopfner, F.; Koeglsperger, T.; Respondek, G.; Schweyer, K.; et al. Four-Repeat Tauopathies. Prog. Neurobiol. 2019, 180, 101644.
- Ganguly, J.; Jog, M. Tauopathy and Movement Disorders—Unveiling the Chameleons and Mimics. Front. Neurol. 2020, 11, 1359.
More