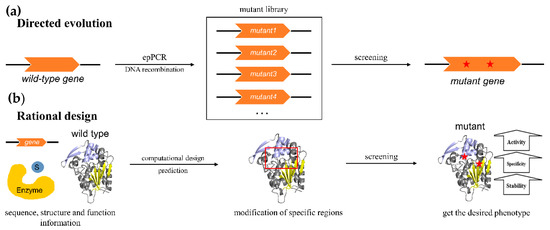
The family of α/β hydrolases is one of the largest known protein families, including a wide range of members such as epoxide hydrolases, dehalogenases, hydroxynitrile lyases, fungal lipases, amidases, dienelactone hydrolases, haloperoxidases, acetylcholine esterases, serine carboxypeptidases, serine carboxypeptidase-like acyltransferases and other enzymes with distinct functions. Although many natural enzymes have been screened as biocatalysts with excellent performance, most of them are still unable to meet the needs of industrial applications. Low catalytic activity, thermostability, and enantioselectivity under complex and harsh industrial process conditions are still the main limitations for the large-scale application of natural enzymes. With the development of protein engineering technology, functional improvements have been achieved for existing α/β hydrolases, specifically in key enzyme characteristics such as their enantioselectivity and stability, in order to tailor these enzymes for specific industrial applications.
- chiral compound
- α/β hydrolase
- catalytic mechanism
1. Structure and Catalytic Mechanism of α/β Hydrolase
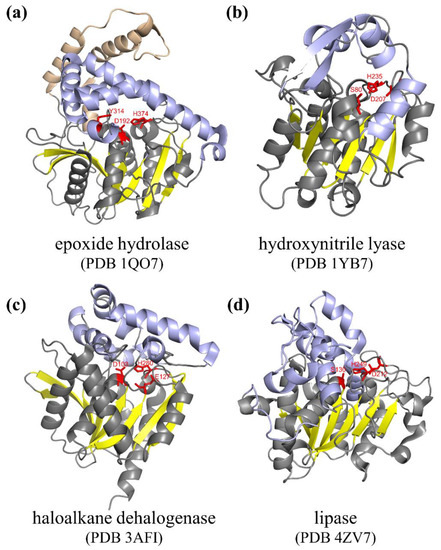
2. Engineering of Natural α/β Hydrolases
| Type | Enzyme | Source | Mutation Site | Reaction/Effects | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Epoxide hydrolase | Alp1U | Streptomyces ambofaciens | W187F/Y247F | Regioselective nucleophilic attack at C-2 of fluostatin C | [11] | [56] | |||||
| Alp1U | Streptomyces ambofaciens | Y247F | Highly regioselective attack at C-3 of fluostatin C | [11] | [56] | ||||||
| Am | EH | Agromyces mediolanus | ZJB120203 | - | Hydrolysis of ( | R | )-ECH to enantiopure ( | S | )-ECH | [12] | [49] |
| An | EH | Aspergillus niger | - | Hydrolysis of epoxides to the more water-soluble and usually less toxic diols | [7] | [22] | |||||
| An | EH | Aspergillus niger | A217L | Improvement in enantioselectivity | [13] | [57] | |||||
| An | EH | Aspergillus niger | A217V | Increase of the activity to allyl glycidyl ether | [13] | [57] | |||||
| An | EH | Aspergillus niger | A217C | Increase of the activity towards allyl glycidyl ether and styrene oxide | [13] | [57] | |||||
| Ar | EH | Agrobacterium radiobacter | AD1 | T247K/I108L/D131S | Improvement of activity, enantioselectivity, and thermostability | [14] | [58] | ||||
| Au | EH2 | Aspergillus usamii | E001 | - | Resolution of racemic styrene oxide | [15] | [47] | ||||
| Au | EH2 | Aspergillus usamii | A214C/A250I | 12.6-fold enhanced enantiomeric ratio toward | rac | -styrene oxide | [16] | [59] | |||
| Au | EH2 | Aspergillus usamii | R322V/L344C | High enantioselectivity towards | rac- | ortho-trifluoromethyl styrene oxide | [17] | [60] | |||
| EchA | Agrobacterium radiobacter | AD1 | I219F | Enhanced enantioselectivity for styrene oxide | [18] | [61] | |||||
| EchA | Agrobacterium radiobacter | AD1 | L190F | Enhanced activity for styrene oxide | [18] | [61] | |||||
| Gm | EH3 | Glycine max | - | Enantioconvergent hydrolysis of | rac | -epoxides with high enantiopurity and yield | [19] | [48] | |||
| SgcF | Streptomyces griseus | IFO 13 350 | W236Y/Q237M | 20-fold increased activity toward ( | S | )-styrene oxide to yield an ( | S | )-diol | [20] | [62] | |
| Sibe-EH | metagenomes | - | Desymmetrization of | cis | -2,3-epoxybutane producing the (2 | R | ,3 | R | )-diol | [21] | [52] |
| CH65-EH | metagenomes | - | EH activity toward a broad range of substrates and with high thermostability |
[21] | [52] | ||||||
| Vr | EH1 | Vigna radiata | - | Enantioconvergent hydrolysis of | p | -nitrostyrene oxide | [22][23] | [44,45] | |||
| Vr | EH2 | Vigna radiata | - | Enantioconvergent hydrolysis of | p | -nitrostyrene oxide | [23] | [45] | |||
| Vr | EH3 | Vigna radiata | - | High and complementary regioselectivity toward styrene oxides and high enantioselectivity toward | o | -cresyl glycidyl ether | [24] | [46] | |||
| Esterase | PFE | Pseudomonas fluorescens | replacement of a loop (A120-V139) with the corresponding element (P132-Y152) of the epoxide hydrolase EchA | Conversion of an esterase into an epoxide hydrolase towards | p | -nitrostyrene oxide | [25] | [63] | |||
| Rh | Est1 | Rhodococcus | sp. ECU1013 | circular permutation mutants with G20/T19, S22/N21, and G24&A23 as new termini, respectively | Improved thermostability | [26] | [64] | ||||
| SABP2 | Nicotiana tabacum | Q221M | Higher stability (6.6-fold half-life) | [27] | [65] | ||||||
| SABP2 | Nicotiana tabacum | G12T/M239K | Switching from an esterase to a hydroxynitrile lyase | [28] | [66] | ||||||
| Haloalkane dehalogenase | DbjA | Bradyrhizobium japonicum | USDA110 | - | Excellent enantioselectivity for α-bromoesters and high enantioselectivity for two β-bromoalkanes | [9] | [24] | ||||
| DbjA | Bradyrhizobium japonicum | USDA110 | H280F | Realized the transhalogenation reaction | [29] | [67] | |||||
| DbeA | Bradyrhizobium elkanii | USDA94 | surface loop-helix transplantation from haloalkane dehalogenase DbjA | Lower stability but increased activity with various halogenated substrates and altered its enantioselectivity | [30] | [68] | |||||
| DhaA | Rhodococcus rhodochrous | E20S/F80R/C128F/T148L/A155P/A172I/C176F/D198W/V219W/C262L/D266F | Increased thermostability (Δ | Tm | 24.6 °C) | [31] | [69] | ||||
| DhaA | Rhodococcus rhodochrous | two mutants containing 13 and 17 mutation sites, respectively | Enantioselective production of ( | R | )-and ( | S | )-2,3-dichloropropan-1-ol, respectively | [32] | [38] | ||
| LinB | Sphingomonas paucimobilis | UT26 |
E15T/A53L/A81K/F169V/A197P/D255A/A247F | Increased thermostability (Δ | Tm,app | 23 °C) | [33] | [70] | |||
| Hydroxynitrile lyase | Hb | HNL | Hevea brasiliensis | L121Y | Improved activity on an unnatural substrate mandelonitrile | [34] | [71] | ||||
| Hb | HNL | Hevea brasiliensis | T11G/E79H/K236G | Lower hydroxynitrile lyase activity and higher esterase-specific activity | [35] | [72] | |||||
| Lipase | CALB | Candida antarctica | - | Kinetic resolution of racemic alcohols and amines or desymmetrization of diols and diacetates | [10] | [25] | |||||
| CALB | Candida antarctica | a circular permutated variant of CALB with 283 /282 as the new termini | Higher catalytic activity (2.6- to 9-fold) for trans and interesterification of the different substrates | [36] | [73] |