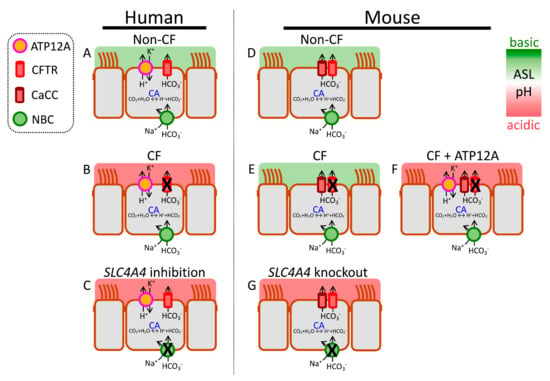
differ between human and mouse airways. Models show key transport mechanisms that determine pH
in human and mouse airway epithelia. Left panel (human): (
despite intact apical CFTR channels. Right panel (mouse): (
) a model of non-CF (wild type) mouse airway epithelium. Note the absence of ATP12A and the expression of non-CFTR (CaCC) HCO
in CF mice, providing one explanation for lack of spontaneous airway disease. However, exogenous ATP12A expression increases H
mice phenocopy human CF. For simplicity, only the chief acid–base transport mechanisms controlling pH
are shown.
-ATPase, which may also influence the movement of acid–base equivalents into or out of ASL. See legend and text for details. CA = carbonic anhydrase.
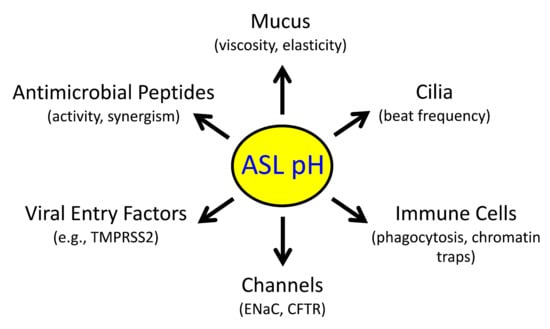
2.1. Reduced pH
ASL
Disrupts Host Defenses
The normal pH
ASL is mildly acidic (e.g., 6.9–7.1 in human lower airways) relative to the interstitial fluid [7.4], though it varies considerably between individuals as well as between studies
[1][27][28][1,36,37]. Recent reviews provide excellent tables summarizing pH
ASL variability between studies, including wild type versus CF
[27][28][36,37]. Absolute pH
ASL measurements are influenced by technique, model system and airway region. It is also interesting to point out that pH is measured on log scale and a 3/10 increase in pH reflects a doubling of [H
+].
Notwithstanding challenges involved in comparing results between studies, recent reports have identified several genetic changes in humans and/or animal models that alter pH
ASL (
Figure 2). These studies provide critical insights into acid–base transport mechanisms that control pH
ASL and thus airway host defenses.
In humans with CF, the loss of CFTR-mediated HCO
3− secretion leaves H
+ secretion unbalanced, and hence lowers pH
ASL [34][51]. An abnormally acidic pH
ASL impairs mucociliary clearance, antimicrobial peptide activity against inhaled pathogens, and phagocytic cell function
[17][18][21][23,24,27]. Raising pH
ASL at least partially rescues these impairments and identifies epithelial acid–base transporters as potential therapeutic targets.
The involvement of pH
ASL in airway host defense has also been tested by disrupting acid–base transporters other than CFTR. For instance, inhibiting basolateral NBC activity in human airway epithelia with normal CFTR channels lowers pH
ASL [35][52]. Interestingly,
CFTR disruption in mice fails to produce spontaneous airway disease
[36][37][53,54]. This is partly due to increased expression of non-CFTR HCO
3− transporters and lack of expression of the non-gastric H
+-pump (ATP12A) in murine airways (see below)
[38][55].
Mutations in
carbonic anhydrase isoform 12 (
CA12) also phenocopy the loss of CFTR channel activity in human airways
[39][56]. People with
CA12 mutations show chronic coughs, airway colonization, bronchiectasis, and elevated sweat [Cl
−]. Interestingly, CA12 localizes at the apical membrane of airway epithelia. This expression pattern is physiologically relevant because proximal airway CO
2 concentration fluctuates during tidal breathing
[40][57]. Thus, pH
ASL rises during inhalation as airway lumen fills up with inhaled air (low CO
2 concentration) but falls during exhalation as alveolar gases (higher CO
2 concentration) enter the airways. Tidal pH
ASL oscillations enhance the epithelial host defense against bacteria.
ASL buffers resist changes in pH
ASL when H
+ ions are added or removed. The main ASL buffer is HCO
3− [41][58]. Accordingly, in Calu-3 epithelia, forskolin stimulation increases and CFTR knockdown decreases buffering capacity of apically secreted fluid. Mucins are negatively charged molecules that can also bind H
+ and thus contribute to ASL buffering
[42][43][59,60]. This effect is dependent on mucin concentration. Mucus accumulation lowers the amplitude of ventilatory pH
ASL oscillations
[40][57], and dampened oscillations reduce antibacterial host defense, thus providing a potential mechanism by which mucus accumulation may increase susceptibility to respiratory infections.
2.2. pH
ASL
Changes with CF Airway Disease Progression
In vitro studies in human airway epithelia show that pH
ASL is abnormally acidic in CF
[34][38][44][51,55,61], but in vivo studies reveal mixed results. Some studies show a lower pH
ASL in CF
[45][46][62,63], whereas others report no difference between CF and non-CF individuals
[47][48][64,65]. This discrepancy is intriguing, given that CFTR-mediated HCO
3− secretion is decreased in both in vitro and in vivo studies.
Most babies with CF develop airway inflammation over the first year of life
[49][50][51][52][53][66,67,68,69,70]; in some cases, they also develop respiratory infections. Inflammation may induce ASL alkalinization through CFTR-independent mechanisms, and thus conceal the loss of CFTR-mediated HCO
3− secretion. Studying initial stages of human CF airway disease might help separate effects due to CFTR loss from those due to inflammation. As inflammation develops early in CF, this approach requires studying ASL from babies soon after birth. In one small pilot study, pH
ASL in newborn CF babies (<four weeks of age) was lower compared with non-CF babies
[54][71].
2.3. Inflammatory Cytokines Regulate pH
ASL
In the absence of rigorous in vivo assessments, the effects of inflammation on pH
ASL may be investigated in vitro by applying CF-relevant inflammatory stimuli to primary cultures of differentiated human airway epithelia. The cytokine interleukin-17 (IL-17) is an evolutionarily conserved molecule that drives neutrophilic airway inflammation
[55][56][80,81]. In targeting the airway epithelium, IL-17 acts synergistically with other cytokines such as tumor necrosis factor-α (TNFα), IL-1β, etc.
[57][82]. These proinflammatory molecules are increased in established CF airway disease
[58][59][83,84].
2.4. H
+
Secretion
Inflammation may alter acid secretion into ASL. One study reported increased expression of ATP12A in CF airways with established disease
[60][88]. Exposure of airway epithelia to IL-4 or IL-13 also increased ATP12A expression, and thus H
+ secretion
[61][62][47,86]. TNFα exposure had a similar effect, but IL-17 alone or combined IL-17/TNFα did not change H
+ secretion
[31][40]. Importantly, inhibiting ATP12A with apical ouabain decreases H
+ secretion, lowers ASL viscosity, and increases bacterial killing
[34][38][62][51,55,86]. Efforts to identify safer agents that reduce ATP12A activity are underway.
Loss of CFTR function also affects distal (small) airways. Small airway epithelia lack ATP12A, but instead use V-ATPase to secrete H
+. Effects of inflammation and infection on small airway H
+ secretion remain relatively unexplored.
P. aeruginosa infection may reduce V-ATPase-mediated H
+ secretion
[63][64][89,90], or acidify ASL via apically expressed monocarboxylate transporter 2, a H
+-lactate cotransporter
[65][91]. Interestingly, Li et al. showed that pH
ASL regulates membrane localization of V-ATPase in porcine small airway epithelia
[32][41]. At neutral pH
ASL (7.4), the V-ATPase subunit ATP6V0D2 localizes in the apical membrane of small airway secretory cells.
2.5. HCO
3−
Secretion
2.5.1. CFTR-Mediated HCO3− Secretion
Several inflammatory cytokines have been shown to alkalinize pH
ASL. IL-17/TNFα, IL-4, IL-13, and IL-1β increase CFTR expression and activity, and increased CFTR-mediated anion secretion improves respiratory host defenses
[31][61][66][67][40,47,92,93]. In CF epithelia, this component of host response is missing due to mutated, non-functional CFTR proteins. In contrast to above-mentioned cytokines, TGF-β reduces CFTR expression and transport activities in airway epithelia
[68][69][94,95]. In vivo effects of inflammation are likely to be complex given that several cytokines elevated in CF airways target airway epithelium, alter HCO
3− transport, and thus modify pH
ASL.
2.5.2. Non-CFTR-Mediated HCO3− Secretion
An array of cytokines (IL-17/TNFα, IL-4, and IL-13) induce non-CFTR HCO
3− secretion across airway epithelia
[30][61][66][70][71][39,47,85,92,96]. This is achieved through pendrin, an apical Cl
−/HCO
3− exchanger, encoded by the gene
SLC26A4. Several aspects of this transport process are noteworthy. First, pendrin is minimally expressed in airway epithelia under basal conditions but is strongly upregulated by inflammatory cytokines. Second, pendrin is an electroneutral exchanger that does not mediate net anion secretion or absorption or change membrane potential. Third, in the absence of functional CFTR channels, pendrin alone can drive ASL alkalinization, though greater alkalinization is achieved with pendrin plus CFTR
[30][31][39,40]. Fourth, some reports indicate structural or functional interactions between CFTR and pendrin, resulting in the increased activity of both
[70][72][85,97].
2.5.3. Paracellular HCO3− Shunt
In addition to secretion by airway epithelial cells, HCO
3− can also move between the cells. Very few studies have explored the contribution of the paracellular pathway to pH
ASL, though it is often mentioned in the context of inflammation. A recent report showed that the paracellular pathway is as permeable to HCO
3− as it is to Cl
− [73][98]. Under basal conditions, pH
ASL (6.6) is lower than the pH of the interstitial fluid (7.4) and the paracellular HCO
3− flux is towards the lumen; however, the paracellular HCO
3− flux decreases or even reverses as pH
ASL approaches or rises higher than the pH of the interstitial fluid. The paracellular pathway thus acts as a HCO
3− shunt that may oppose increased cellular HCO
3− secretion in inflamed airway epithelia
[74][99]. Whether paracellular HCO
3− permeability can be modulated to support ASL alkalinization is an interesting question and requires further investigation.
2.6. Other Regulatory Mechanisms
Inflammatory cytokines regulate diverse cellular mechanisms involved in HCO
3− secretion. In addition to changes in apical HCO
3− transporters, cytokines such as IL-17/TNFα, IL-13, or IL-4 also increase transcripts of several carbonic anhydrase and NBC isoforms
[30][61][39,47]. Additional cytoplasmic mechanisms that regulate epithelial HCO
3− secretion also change in the presence of cytokines. The WNK (with-no-lysine [K]) kinases are master-regulators of pancreatic HCO
3− secretion
[75][100]. As reported recently, these kinases also control HCO
3− secretion across CF and non-CF airway epithelia
[76][101]. Secretory cells, key HCO
3− secreting cells in airway epithelia, express two WNK isoforms, WNK1 and WNK2. Reducing WNK kinase activity increases HCO
3− secretion, raises pH
ASL, and enhances CF epithelial host defenses. At a mechanistic level, WNK kinases regulate intracellular [Cl
−] through their control of the basolateral Na
+-K
+-2Cl
− cotransporter (NKCC1). Inhibiting WNK lowers intracellular [Cl
−], which in turn may act as a signaling ion to stimulate HCO
3− transport
[77][78][102,103]. It is of note that combined IL-17/TNFα reduce WNK2 expression and raise pH
ASL and inhibiting residual WNK kinase activity further alkalinizes ASL.