Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Charandeep Singh and Version 2 by Dean Liu.
The retina is one of the most metabolically active organs in the body. Although it is an extension of the brain, the metabolic needs of the retina and metabolic exchanges between the different cell types in the retina are not the same as that of the brain. Retinal photoreceptors convert most of the glucose into lactate via aerobic glycolysis which takes place in their cytosol, yet there are immense numbers of mitochondria in photoreceptors.
- metabolism
- ROP
- retinal metabolism
1. Introduction
The retina is formed by very well-organized layers of different types of cells with close metabolic symbiotic exchanges between these cell types (Figure 1). Retinal pigmented epithelium (RPE) serves as a barrier between the retina and choriocapillaris. Based on the blood supply distribution to the retina, retinas can be classified as vascular or avascular. The vascular retina is further subdivided in euangiotic or holangiotic pattern (humans, pigs, dogs), merangiotic (a smaller part of the retina like the rabbit), and paurangiotic (minute and restricted to the direct neighborhood of the optic disc, like the horse and the guinea pig). The avian retina is completely avascular (anangiotic pattern) [1]. Vascularized retinae have a dual blood supply mechanism, supplied with oxygen and nutrients through the retinal blood supply in addition to the choroidal blood supply. Blood vessels in the vascularized retina have tight junctions which serve as blood-retinal barrier to protect the retina from pathogens or toxins [2]. Unlike retinal vasculature, choroidal vasculature is fenestrated, but the retinal pigmented epithelium has tight junctions to form a blood-retinal barrier between the choroidal blood supply and the retina [3].
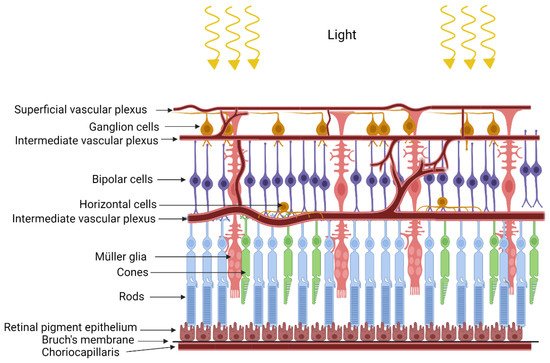
Figure 1. Structure of the retina.
2. Retinopathy of Prematurity
Retinopathy of prematurity is a neurovascular retinal defect in prematurely born infants. Supplemental oxygen is needed to prevent mortality in the prematurely born infants with underdeveloped lungs [4][28]. On one hand, supplemental oxygen prevents motility, but on the other hand it suppresses development of the retinal blood vessel leading to hypovascularized retina in the phase I of ROP. The underdeveloped vasculature in phase I in the developing retina causes local hypoxia, due to limited supply of blood to the inner retina, leading to neovascularization in phase II of ROP [5][6][7][8][29,30,31,32]. There are several different animal models of oxygen-induced retinopathy (OIR) which can recapitulate neuro-vascular abnormalities seen in ROP [9][33]. Of all the models, mouse and rat models are used more often because they are economical, have large litter sizes, and are reproducible and robust models. The mouse OIR model has been used for a majority of the animal metabolic studies. All the animal models have the same underlying principle, i.e., animals are first subjected to a hyperoxic treatment which ceases blood vessel growth in the retina, followed by a return to normoxic room air causing local hypoxia in the retina which triggers retinal neovascularization [9][10][33,34]. Phase I and Phase II cause opposite effects on retinal vasculature development, and both these phases are expected to show complementary metabolic changes. Under normal development, new blood vessels are formed by proliferating and migrating endothelial cells. Proliferating endothelial cells have very high glutamine needs, and this need is fulfilled by the Müller cells [11][12][35,36]. Müller cells are the sole producers of retinal glutamine [13][37]. Baseline glutamine lyase (GLS) activity in the endothelial cells is higher than in any other cell type, implying that they are major consumers of glutamine. Although endothelial cells use glycolysis for their energy needs, they consume glutamine during development, which implies glycolysis in the endothelial cells is uncoupled from TCA. In utero, blood vessels develop in a relatively hypoxic environment. Blood vessel development needs a hypoxic environment, where HIF-dependent pathways such as VEGF gradients, lactate gradients derived from the retinal neurons and Müller cells, play a central role in development [13][14][37,38]. All three, endothelial cells, neurons, and Müller cells, are dependent on each other for their metabolic needs during retinal vasculature development in the retina. In ROP and OIR models, hyperoxia degrades all isoforms of HIF-α by prolyl hydroxylase domain protein (PHD) dependent hydroxylation, a post-transitional modification, which requires molecular oxygen as one of the substrates [4][15][28,39]. Hydroxylation of HIF is followed by ubiquitination, which marks it for proteasomal degradation, whereas hypoxia stabilizes HIF and is beneficial for during the retina development [4][28]. Hypoxic preconditioning has been demonstrated to be beneficial in angiogenesis, to prevent neurons from ischemic injury, and to protect photoreceptors from light damage [16][17][18][40,41,42]. An alternative strategy to stabilize HIF, by PHD inhibition using pharmacological inhibitor dimethyloxallyl glycine (DMOG), has been applied in cardio-protection in ischemic injury models [19][43]. Hepatic HIF stabilization by PHD inhibition either by DMOG or FG-4592 has been demonstrated to be protective against phase I of OIR in rat and mouse models [20][21][22][44,45,46]. Hepatic HIF1 knockout (KO) mice are not protected against OIR when treated with DMOG, implying that DMOG acts via hepatic HIF dependent pathways [21][45]. To understand the metabolic basis of OIR and protection in HIF stabilized condition, rwesearchers performed an untargeted metabolite profiling of polar metabolites extracted from the retina or the plasma of the P10 old noromoxic, hyperoxic, and hyperoxic mice treated with HIF stabilizer FG-4592 [4][28]. ResWearchers found increased serine/one-carbon pathway flux in hyperoxic mice treated with FG-4592. Serine/one-carbon pathway provides biosynthetic precursors such as nucleic acids, NADPH for lipogenesis, and s-adenosylmethonine for methylation reactions to the developing cells in the retina. In addition, researcherswe also found systemic upregulation of proline and urea cycle metabolites in OIR mouse models treated with HIF stabilizer FG-4592. Moreover, using 13[C]6 glucose, rwesearchers demonstrated that the retinal explant from P10 age mice were unable to produce their own serine or glycine, and retinal serine at this age is derived from circulation. Inhibition of serine/one-carbon increased avascular area in the retinas treated with FG-4592 as compared to controls [4][28]. Paris et al. analyzed metabolic difference in the ocular metabolome (whole eye) of the OIR model mice vs. nomoxic controls, in the phase II at P12, P14, and P17. Arginine and interconversion to proline, urea cycle, and β-oxidation were also demonstrated to be upregulated in the whole eye metabolome of the OIR phase II mouse model, whereas purine pathway metabolites were found to be downregulated [23][47]. These findings together reflect downregulation of biosynthetic metabolites necessary for normal growth of the retina in phase I and upregulation of these pathways in proliferative phase II of OIR. Tomito et al. analyzed metabolites in the vitreous humor from proliferative diabetic retinopathy patients and retinal metabolites from P17 old OIR model hypoxic phase II of the mouse model. They found decreased levels of creatine and increased levels of glycine, in both the Proliferative Diabetic Retinopathy (PDR) patient vitreous samples and hypoxic P17 mouse retina. Creatine supplementation reduced OIR-linked neovascularization (NV) in the mouse model via downregulation of Vegf-a and Pdgf-b [24][48]. The creatine production pathway involves production of guanidinoacetate from arginine and glycine. By the action of Guanidinoacetate N-methyltransferase (GAMT) enzyme, guanidinoacetate and s-adenosylmethioine (SAM) produce creatine. The SAM required for this step is produced through carbon donated from serine in the one-carbon pathway. In the absence of SAM or inhibition of GAMT, one would expect to see substrates upstream of the enzyme, such as guanidinoacetate, glycine, and arginine, to accumulate. Zhou et al. performed a time-series comparative untargeted metabolomics analysis on the retinas from OIR model mouse and room-air controls at P12 (postnatal day 12), P13, P17, and P42. Many amino acids were found to be perturbed, confirmed by a targeted amino acid measurement. They found downregulation of many amino acids in P12 OIR mouse retinas which were non-significant, and after removal of mice to room air from P13-P42, amino acids were statistically significantly upregulated [25][49]. In a separate study, Zhaou et al. also screened plasma metabolome of the treatment requiring ROP patients and aged-matched controls. They found significant differences in the “aminoacyl-tRNA biosynthesis” and “protein digestion and absorption”, and these pathways were enriched via KEGG analysis of the untargeted metabolomics data. Using variable importance in projection in multivariate statistical analysis, they found 29 and 23 metabolite pool size differences in positive and negative ionization modes, respectively. However, using univariate comparisons of the metabolomics data, they found only 11 metabolites in positive and 13 metabolites in negative ionization modes to be different with the univariate comparison [26][50]. All these recent studies indicate perturbation in amino acid metabolism, specifically serine/one-carbon metabolism, in arginine and proline, and nitrogen metabolism in the ROP or OIR. Of these metabolic pathways, arginine metabolism to ornithine by arginase has been studied earlier in a neurodegeneration context in retinal OIR models. Mitochondrial isoform of arginase, arginase 2, has been shown to be involved in apoptosis of neurons in the mouse OIR model [27][51]. Ornithine, produced by arginase reaction, is utilized to produce polyamines and can additionally be used for proline synthesis, both of which are important biomass precursors. The mechanism underlying arginase 2 deficiency being beneficial in OIR models has been shown to involve excessive production of polyamine and oxidation of polyamine products to their precursor generating toxic side products. Arginase 2 activity has been localized mainly to horizonal cells in the retina. Narayanan et al. demonstrated increased expression of polyamine oxidation enzyme spermine oxidase (SMO) in response to hyperoxia. Toxic side products from polyamine breakdown, such as acrolein, 3-acetaminopropanol, 3-aminopropanol, and H2O2, have been implied to underlie the neurotoxic apoptotic activity in the OIR retina [27][51]. RWesearchers demonstrated that hyperoxia downregulates proliferation of human primary retinal endothelial cells in the culture. ResearchersWe have also seen upregulation of polyamine degradation enzymes SMOX and PAOX, and decreased levels of putrescine in hyperoxic treated primary endothelial cells. Researchers We demonstrated downregulation of Myc in response to hyperoxia [28][52]. Myc and HIF have very overlapping control over biosynthetic pathways. Ornithine decarboxylase is one of the Myc targets, and ODC has been demonstrated to peak at the G1-S and G2-M phase of the cell cycle [29][53]. This demonstrates that metabolic signaling by polyamine metabolites triggers cell cycle arrest in endothelial cells. In addition, reswearchers also demonstrated that hyperoxia alters glutamine metabolism in retinal Müller cells. Hyperoxia blocks entry of glycolytic flux into TCA and in response to which Müller cells start to use glutamine for their anaplerotic needs [30][54]. Cultured primary and immortalized Müller cells exposed to hyperoxic conditions lose this property and can no longer assimilate ammonium into glutamine [30][54]. Müller cells are the sole producers of glutamine, and in hyperoxic conditions rwesearchers sa saw a reversal of this function to glutamine utilization rather the production. These alterations probably explain why, in many studies, urea cycle metabolites such as arginine, ornithine, and proline were found to be altered. Other than the central carbon metabolism discussed above, polyunsaturated fatty acids, arachidonic acid, and Docosahexaenoic Acid (DHA) have been shown to be low in the blood of preterm infants who develop ROP [31][32][33][34][55,56,57,58]. The mouse model of OIR treated with DHA improves, and Arachidonic acid (AA) exacerbates, neovascularization [35][36][59,60]. Altogether, all the studies point toward a systemic metabolic imbalance underlying ROP development, and a systemic biochemical multi-organ metabolic model is much needed to understand and find treatment for ROP. Biochemical alterations seen in OIR/ROP have been summarized in the Figure 2.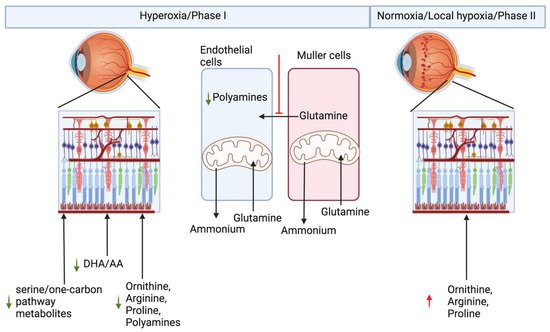
Figure 2. Metabolic basis of retinopathy of prematurity.
3. Diabetic Retinopathy
Diabetic retinopathy is an example of systemic pathogenesis, where uncontrolled systemic hyperglycemia underlies the damage to the retinal blood vessels. Diabetic vascular retinopathy can be divided into two sub-types based on progress in the disease development. Early stage is referred to as non-proliferative DR, which is characterized by blood vessel damage and loss. Severe microvascular loss to the blood vessels supplying nutrients to the retina leads to neuronal damage and causes local hypoxia in the retina. This local hypoxia initiates the more aggressive stage of the DR, referred to as proliferative DR, in which neovascularization takes place. Sustained hyperglycemia leads to buildup of sorbitol by the action of aldose reductase [37][72]. Sorbitol, a sugar alcohol or polyol, causes osmotic stress in the retina, thereby further damaging blood vessels in the retina. Sustained systemic hyperglycemia also results in non-enzymatic spontaneous glycation of proteins which are referred to as advanced glycation end products (AGE). Diabetes leads to the breakdown of the blood-retinal barrier, leading to leaky blood vessels. Leaky vasculature allows accumulation of fluid in the retina and consequently leads to macular edema, causing vision loss in the macular region of the retina. Apart from the vascular component of the disease, other cells such as Müller cells contribute to the neurovascular component of the pathology. Diabetes induces GFAP expression in the Müller cells which leads to gliosis [38][73].
There are ample metabolic studies on DR patients published lately, and it is out of the scope of this manuscript to discuss them all in this review. Only a few of the latest research articles have been discussed here. Tomita et al. compared vitreous metabolome from PDR patients to non-diabetic controls. They found increased concentrations of lactate, pyruvate, allantoin, and proline in PDR patients compared to non-diabetic controls. Pathway enrichment analysis demonstrated changes in serine, glycine, proline, and arginine pathways. They also found increased concentrations of glycine and decreased concentration of downstream product creatine [24][48]. Yun et al., measured metabolic changes in the plasma of non-proliferative and proliferative DR and compared it the non-diabetic controls. They found 16 metabolites which separated DR patients from non-diabetic controls. Kynurenine, tryptophan, and dimethyl arginine were demonstrated to be markers differentiating DR progression in the patients [39][74]. Zhu et al. compared the plasma metabolome of PDR patients to that of the DR patients with ≥10 years of diabetes without DR retinopathy [40][75]. They found four reliable PDR plasma biomarkers: fumaric acid, acetic acid, cytidine, and uridine [40][75]. Another study by Sumarriva et al. compared plasma metabolome of Non-proliferative diabetic retinopathy (NPDR), PDR, and non-diabetic controls. They found alterations in “arginine and citrulline-related pathways” in DR compared to non-DR controls. Additionally, Carnitine was found to be different between NPDR and PDR groups, implying alterations in fatty acid metabolism with disease progression [41][76]. Peters et al. performed a targeted screening of arginine, asymmetric dimethylarginine, proline, ornithine, arginosuccinate, and citrulline, using isotope dilution mass-spectrometry in the plasma metabolome of diabetic controls, NPDR and PDR. All four metabolites were found to be increased in the citrulline, arginine, arginosuccinate, ADMA, and ornithine in DR vs. diabetic controls. However, after multiple testing and thorough statistical adjustments for false positives, only arginine and citrulline were found to be elevated in type 2 DR patients [42][77]. Sun et al. compared plasma metabolome of DR and PDR patients and found differences in four metabolites: N-acetyltryptophan, glutamate, pseudouridine, and leucylleucine in these patients [43][78].
There are many different animal models to study DR. It can be induced by complete or partial removal of the pancreas or damage caused by certain drugs targeting pancreases. DR can also be induced by a diet high in galactose or glucose, but these diet-induced models are used less frequently. Oxygen-induced retinopathy models are also used as a proxy of advanced PDR. There are also genetic models, like Akita (Insulin mutations leading to loss of beta cells and insulin production mimicking type I diabetes), db/db (Leptin Receptor mutation leading to obesity and diabetes), Kimba (trVEGF029) and Akimba (cross between Akita and Kimba). The most used DR models are streptozotocin-induced, db/db and Akita mice. Mora-Ortis et al. performed NMR-based metabolomics of db/db mouse model tissues, including of the eye. They found increased amounts of lipids and glucose in the eyes of db/db mice. Metabolites which were found to be decreased in the eyes of db/db mice were citrulline, alanine, glutamate, GABA, glutamine, hypoxanthine, isocitrate, myo-inositol, phenylalanine, O-phosphocholine, tyrosine, and inosine [44][79]. Patrick et al. used STZ-induced DB rat models and demonstrated changes in NADPH/NADP+, ATP/ADP, ADP/AMP, FADH2, tyrosine, citrulline, glutamine, methionine, leucine, aspartate, xanthine, pyridoxal phosphate, 4-aminobutanoate, riboflavin, 2-phosphoglycerate, PEP, 2-oxoglutarate, glycerone phosphate, sorbitol, acetyl CoA, glyoxylate, succinate, HCO3−, and CO2 [45][80].