1. Overview of Perhexiline
Perhexiline maleate (prescribed as Pexsig or Pexid) was originally developed as an anti-anginal drug in the 1970s (reviewed in
[1][6]). It was an effective treatment for angina, but reports of severe hepatoxicity and neurotoxicity in a subset of patients resulted in reduced global usage by the late 1980s
[2][3][9,10]. Nevertheless, perhexiline continues to be prescribed in several countries, including Australia and New Zealand, where it is approved for treating patients with refractory angina pectoris or patients with angina in whom other therapies are contraindicated
[4][5][11,12]. Other reported clinically beneficial effects include enhancing myocardial energetics in heart failure
[6][13] and hypertrophic cardiomyopathy
[7][14].
Perhexiline, 2-(2,2-dicyclohexylethyl) piperidine, is a small, amphiphilic molecule with a –CH-CH2 carbon chain backbone, two saturated cyclohexane rings and a piperidine ring (
Figure 1). It is a chiral molecule due to asymmetry of the second carbon of the piperidine ring, and the clinical formulation of perhexiline is a racemic 1:1 mixture of (+)-perhexiline and (−)-perhexiline enantiomers. Perhexiline is metabolised by the polymorphic enzyme cytochrome P450 2D6 (CYP2D6). Variable expression of and/or mutations in CYP2D6 can result in highly variable clearance rates, with distinct phenotypes termed ultrarapid, extensive, intermediate and poor metabolisers
[8][15]. Pharmacokinetic studies suggest that the perhexiline enantiomers display stereoselective metabolism, with the clearance rate of (−)-perhexiline greater than (+)-perhexiline
[9][10][11][16,17,18].
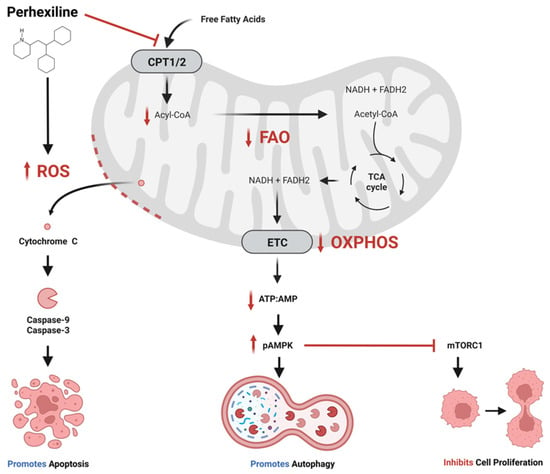
Figure 1. Proposed anti-cancer effects of perhexiline. Perhexiline inhibition of carnitine palmitoyltransferase 1 (CPT1) and CPT2 restricts the entry of free fatty acids into the mitochondrial matrix, thereby inhibiting fatty acid oxidation (FAO). This may limit the production of the electron transport chain (ETC) co-enzymes nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), which would inhibit oxidative phosphorylation (OXPHOS) and the generation of adenosine triphosphate (ATP). A reduction in ratio of ATP to adenosine monophosphate (AMP) activates AMP-activated protein kinase (AMPK) by phosphorylation (pAMPK). pAMPK triggers autophagy, and inhibits cell proliferation by inhibiting mammalian target of rapamycin complex 1 (mTORC1). Additionally, perhexiline increases the level of reactive oxygen species (ROS), which compromises mitochondrial membrane integrity, leading to the release cytochrome c and activation caspases that initiate apoptosis. Created with BioRender.com.
The clinical benefits of perhexiline are derived from its ability to inhibit the mitochondrial enzymes CPT1 and CPT2
[12][13][7,8]. As part of the carnitine system, CPT1 and CPT2 are involved in the translocation of fatty acids across the mitochondrial membranes into mitochondrial matrix, where they undergo FAO (β-oxidation). The oxidation process generates acetyl-CoA, nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2). Acetyl-CoA enters the tricarboxylic acid (TCA) cycle generating more NADH and FADH2, which are co-enzymes in the electron transport chain, and these intermediates are used to generate cellular adenosine triphosphate (ATP). In the cardiac setting, inhibition of the CPTs by perhexiline shifts myocardial metabolism from principally fatty acid toward a greater carbohydrate metabolism, maintaining myocardial production of ATP but requiring lower oxygen consumption
[14][19].
Despite its unique mechanism of action and proven efficacy, the clinical use of perhexiline is limited owing to its narrow therapeutic index and variable pharmacokinetics
[3][15][16][10,20,21]. These factors mean that for long-term clinical use, plasma concentrations of perhexiline must be monitored and maintained within a therapeutic range of 0.15 to 0.60 mg/L (equivalent to ~0.5 to 2 µmol/L)
[17][22]. With monitoring, the risk of serious toxicity associated with long-term perhexiline dosing can be minimised, without abolition of the antianginal effects
[4][17][11,22]. Further, perhexiline toxicity takes several months to develop. Therefore, much higher doses than normally prescribed can be tolerated if perhexiline is administered in cycles, provided there are sufficient breaks between the cycles
[4][17][11,22].
2. Perhexiline: More than Just CPT Inhibition
While perhexiline is widely considered to act by inhibiting CPT and FAO, a number of reports suggest that its anti-cancer effects may be mediated by CPT-independent pathways.
2.1. PI3K/Akt/mTOR
The phosphoinositide 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) signalling pathway coordinates the uptake and utilisation of multiple nutrients, including lipids, glucose, glutamine and nucleotides, facilitating the enhanced growth and proliferation of cancer cells. The pathway is one of the most frequently altered in human cancers. Several studies have demonstrated that perhexiline suppresses the PI3K/Akt/mTOR pathway
[18][19][20][21][24,33,40,44].
PI3K transduces upstream signals from receptor tyrosine kinases to generate critical lipid second messengers that activate downstream signalling effectors such as Akt and mTOR. The conserved serine/threonine-protein kinase, mTOR, belongs to the PI3K family of protein kinases and constitutes the catalytic component of two distinct multiprotein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2)
[22][68]. The mTORC1 complex contains regulatory-associated protein of mTOR (raptor), and the mTORC2 complex contains the rapamycin-insensitive companion of mTOR (rictor). The mTORC1 catalyses the phosphorylation of S6 kinase β-1 (S6K) and initiation factor 4E binding protein 1 (4E-BP1). In contrast, mTORC2 initiates phosphorylation of Akt and protein kinase C (PKC), thereby regulating nutrient metabolism, protein synthesis, growth factor signalling, cell growth and migration
[23][24][69,70].
Balgi et al. identified that perhexiline inhibited mTORC1 signalling, as evidenced by decreases in S6K and 4E-BP1 phosphorylation, and induced autophagy in the breast cancer cell line MCF-7 maintained in nutrient-rich conditions
[25][29]. In contrast, perhexiline did not inhibit mTORC2, suggesting that it did not inhibit mTOR catalytic activity, but rather inhibited signalling to mTORC1. Similarly, Rathore et al. demonstrated that perhexiline treatment of an osteosarcoma cell line (NOS1) significantly decreased levels of total mTOR and total RSP6
[20][40]. Nassar et al. demonstrated that perhexiline induced modest inhibition of ERK and Akt phosphorylation in C4-2B prostate cancer cells
[21][44].
Inhibition of the PI3K/Akt/mTOR pathway has been observed with other CPT inhibitors
[26][71]. Combination therapy with etomoxir (an irreversible inhibitor of CPT1A), and orlistat (an irreversible inhibitor of lipases and fatty acid synthase) resulted in a synergistic decrease in viability of prostate cancer cell lines (LNCaP and VCaP) and patient-derived benign and prostate cancer cells. These effects were associated with decreased mTOR signalling, decreased androgen receptor expression, and increased apoptosis. Knockdown of
CPT1A expression in LNCaP cells decreased oxidation of the fatty acid palmitic acid (C16:0), increased sensitivity to etomoxir, inactivated Akt and activated apoptosis.
2.2. ErbB3 (HER3)
The ErbB family of proteins consist of four structurally related receptor tyrosine kinases: ErbB1 (HER1, EGFR), ErbB2 (HER2, Neu), ErbB3 (HER3) and ErbB4 (HER4). Excessive ErbB signalling is associated with the development of various solid tumours and is associated with poor patient outcomes. Heterodimerisation of HER3 with HER2 activates the PI3K/Akt/mTOR pathway
[27][72], and HER3 knockout impairs the ability of HER2 to induce tumour formation
[28][73].
Ren et al. demonstrated that perhexiline inhibited the activation of HER3 and the proliferation of HER3+ breast cancer cell lines in vitro and in xenografts
[18][24]. The perhexiline treatment induced rapid HER3 internalisation and a reduction in phosphorylated HER3 (pHER3), an active form for HER3 signalling. In contrast, expression of other ErbB family members (EGFR and HER2) were unaffected by perhexiline treatment. Etomoxir had no effect on HER3 localisation, providing limited evidence that perhexiline may operate through mechanisms independent of CPT inhibition. Furthermore, perhexiline inhibited phosphorylation of Akt and ERK1/2, downstream effectors of HER3 signalling. Similarly, Zhu et al. demonstrated that perhexiline inhibited phosphorylation of Akt and other downstream HER3 activation markers in
NKX2-8-deleted (
NKX2-8+/−) CAOV3 and OVCAR3 epithelial ovarian cancer (EOC) cells
[19][33].
2.3. FYN
Tyrosine-protein kinase Fyn (FYN) is a membrane-associated non-receptor tyrosine kinase belonging to the Src family of kinases. It is aberrantly expressed in various cancers contributing to multifaceted signalling that regulates aspects of tumour progression including cellular differentiation, proliferation, antiapoptotic activity, increased migration and motility
[29][30][31][32][33][74,75,76,77,78]. FYN negatively regulates AMPK activity, thereby promoting cell migration and invasion through AMPK/mTOR-mediated signalling
[33][78]. Furthermore, FYN plays a functional role in modulating redox stress by negatively regulating NAPD oxidases that inhibit production of ROS
[34][79].
Kant et al. investigated the effects of perhexiline on FYN in glioblastoma
[35][34]. Perhexiline induced concentration-dependent cytotoxicity in undifferentiated glioblastoma stem cells (PN19 and MES83), and these were more sensitive than differentiated cell lines (T98G and U251). Induced differentiation of PN19 and MES83 increased resistance to perhexiline. However, in contrast to etomoxir, they observed that 5 µmol/L perhexiline failed to alter the oxygen consumption rate in MES83 and T98G cells and did not modulate intracellular lipid dynamics in MES83, leading to the conclusion that the anti-tumour effects of perhexiline were independent of CPT and FAO inhibition in these cells. To identify alternative perhexiline targets, they used SwissTargetPrediction in silico analysis
[36][80], and identified several high probability molecular targets of perhexiline, including FYN, EGFR and family AG protein-coupled receptors (muscarinic acetylcholine receptor M1, M3-5). Surprisingly, CPT1 was not a reported high-probability target. Expression of FYN was high in the perhexiline-sensitive and low in the resistant cell line T98G, unlike the other top targets. FYN expression was significantly higher in glioblastoma, particularly in the proneural subtype, than in normal brain. Perhexiline treatment of glioblastoma cell lines (PN19, MES83 and U251) inhibited FYN activation, as determined by a time-dependent increase in FYN phosphorylation.
2.4. HES1
Hairy and enhancer of split-1 (HES1) is one of seven members of the Hes gene family (HES1-7) that encode basic helix-loop-helix transcription factors which suppress transcription. HES1 has a central role in NOTCH1-induced leukaemia suggesting that abrogation of HES1 activity in leukemia lymphoblasts could be exploited therapeutically. To identify potential small molecule inhibitors of HES1, Schnell et al. interrogated the Connectivity Map
[37][81], a large collection of genome-wide transcriptional expression data derived from cell lines treated with bioactive small molecules, for compounds with transcriptional signatures that overlapped with that induced by HES1 deletion in NOTCH1-induced T-cell acute lymphoblastic leukaemia (T-ALL)
[38][45]. Perhexiline was identified as a potential therapeutic agent for T-ALL due to its ability to elicit a gene expression signature resembling that induced by HES1 deletion in NOTCH1-induced T-ALL. Perhexiline downregulated HES1 expression in CUTLL1 T-ALL cells in vitro. Notably, perhexiline treatment resulted in a significant anti-tumour response and extended survival in mice bearing NOTCH1-induced T-ALL, without significant detrimental effects on the hematopoietic system.