Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Theera Tongsong.
Fetal heart failure (FHF) is a condition of inability of the fetal heart to deliver adequate blood flow for tissue perfusion in various organs, especially the brain, heart, liver and kidneys. FHF is associated with inadequate cardiac output, which is commonly encountered as the final outcome of several disorders and may lead to intrauterine fetal death or severe morbidity. Fetal echocardiography plays an important role in diagnosis of FHF as well as of the underlying causes.
- echocardiography
- fetus
- cardiac function
- heart failure
1. Fetal Dysrhythmias
It has long been known that persistent fetal dysrhythmia can be associated with FHF. However, in recent times, there has been a decline in such association because of earlier detection and intrauterine treatment before the development of FHF [83][1].
-
Supraventricular tachycardia and atrial flutter; the most common causes in this group, usually associated with tachycardia of greater than 220 bpm
-
AV block with cardiac defects; commonly associated with left atrial isomerism and corrected transposition of the great arteries
-
AV block without congenital heart defects, commonly associated with SSA/SSB (Ro/La) antibodies or idiopathic
-
Accelerated junctional or ventricular rhythm, relatively rare
Supraventricular tachycardia (SVT) (Figure 21) and atrial flutter are the most common causes of fetal tachycardia, accounting for 66 to 90% of all cases and also constituting the most common fetal arrhythmia associated with the evolution of FHF [47,84][2][3]. In SVT, the tachycardia range is approximately 220 to 240 bpm with 1:1 ratio of AV conduction and loss of fetal heart rate variability. Atrial flutter is defined as a rapid regular atrial rate of 300 to 600 bpm, accompanied by a variable degree of AV conduction block, resulting in a slower ventricular rate, approximately 220 to 240 bpm. Ventricular tachycardia presents with ventricular rates of more than 180 bpm in the setting of AV dissociation. Though it can be associated with FHF [85][4], it is a rare cause.
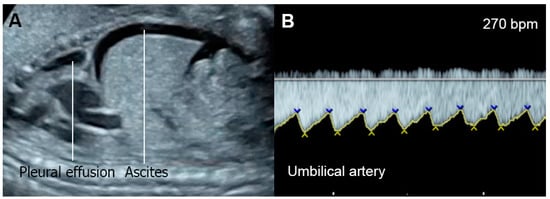
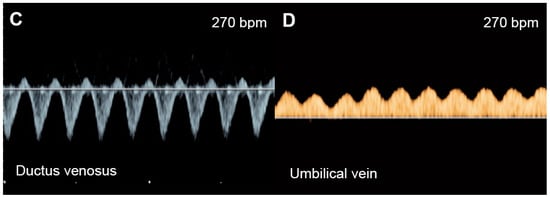
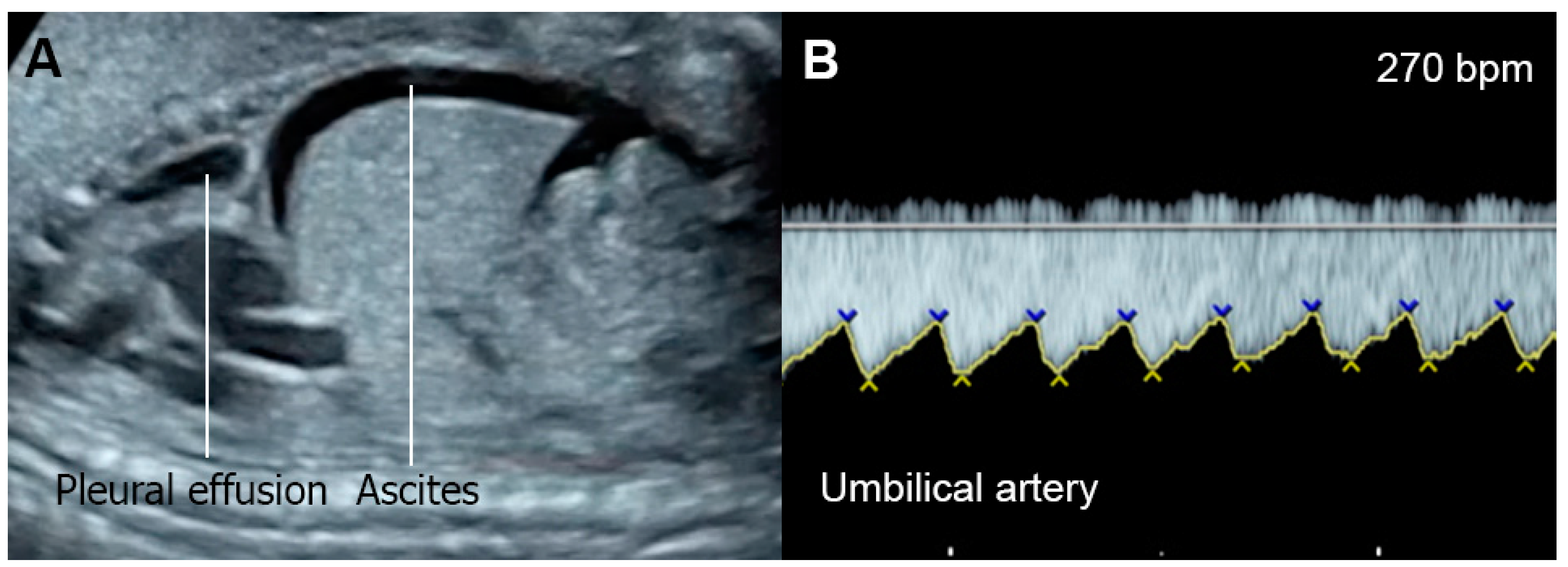
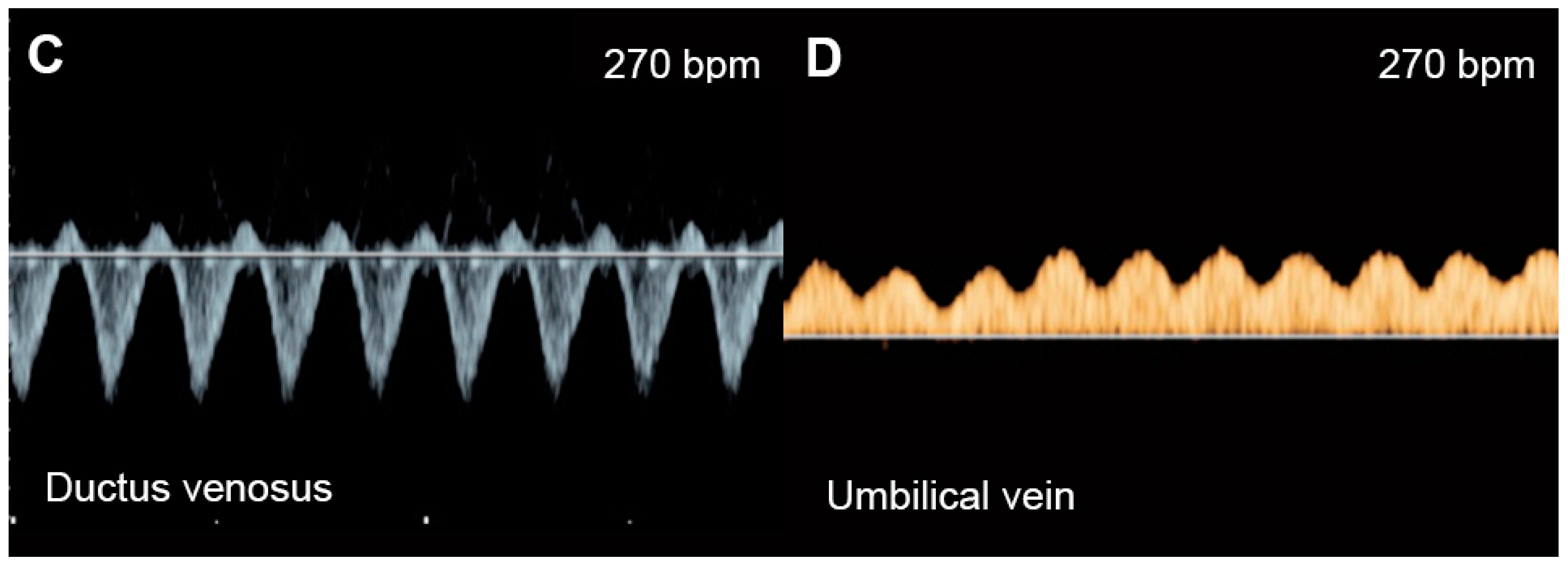
Figure 21. Supraventricular tachycardia: (A) hydropic signs; (B) high end-diastole velocity due to rapid heart rate; (C) ductus venosus Doppler: reversed A-wave, no D-wave due to rapid heart rate; (D) venous pulsations in the umbilical vein.
The mechanisms responsible for heart failure caused by SVT may be associated with increased ventricular and atrial pressures and myocardial energy loss, leading to cardiac dysfunction and culminating in FHF. Very rapid heartbeat directly decreases ventricular filling and ejection time, causing an increase in ventricular and atrial pressures. Gembruch et al. [86][5] demonstrated that in human fetuses with SVT (greater than 210 bpm), systemic venous pressure was significantly increased, as indicated by an increase in the reversal of A-waves of venous Doppler flow patterns in the IVC and the ductus venosus, as a consequence of increasing reversal of flow in atrial systole.
Fetal bradycardia, defined as a sustained fetal heart rate of less than 100 bpm, is commonly caused by sinus bradycardia, blocked atrial bigeminy/trigeminy, or a high-degree AV block. Of the bradycardia group, a complete atrioventricular block (CAVB) (Figure 32) is the most common entity in association with the evolution of FHF. Approximately 50% of CAVB cases occur in association with maternal autoantibodies related to connective tissue diseases; 40–50% of CAVB cases occur in fetuses with congenital heart defects, and the remaining are of unclear etiology (4–10% of cases) [90][6].
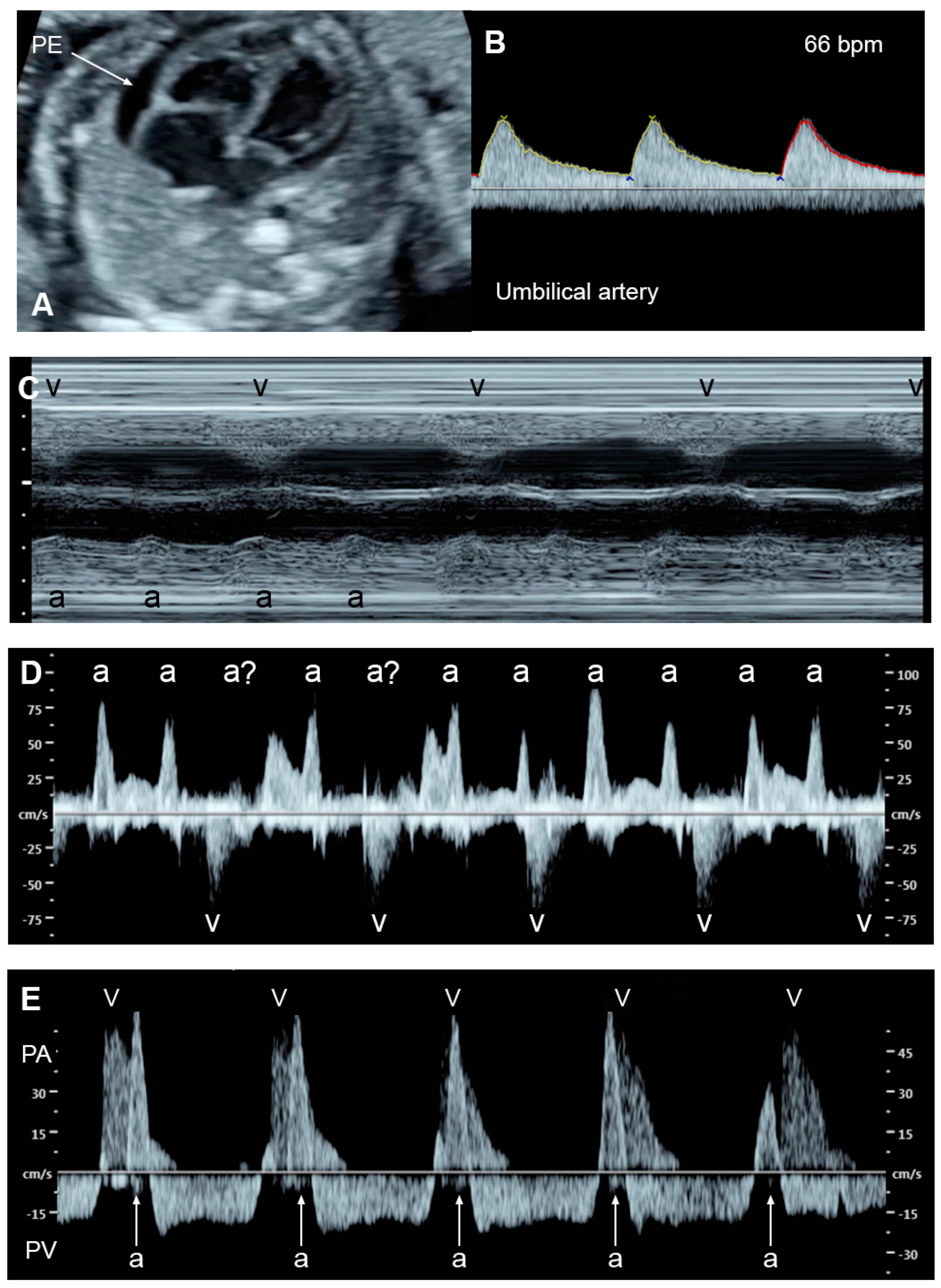
Figure 32. Complete heart block: (A) slightly enlarged heart with pericardial effusion (PE); (B) Ven-tricular rate of 66 bpm in the umbilical artery; (C–E) dissociations between atrial and ventricular contraction with ventricular bradycardia in M-mode (C) cardiac Doppler (D), and PW-Doppler of the pulmonary artery (PA) and pulmonary vein (PV) (E).
Fetuses with an AV block make attempts to increase compensatory ventricular stroke volumes to maintain adequate cardiac output. Nevertheless, some cannot tolerate such excessive workload, culminating in FHF and, subsequently, fetal demise. FHF and fetal demise associated with AV block are more common in cases with a ventricular rate of less than 55 bpm, which increases the risk of fetal demise and should be considered as a poor prognostic factor [90,91][6][7]. Additionally, several studies have shown that fetal heart block in the presence of a structural cardiac defect is associated with a very high perinatal mortality rate [90,92][6][8]. Additionally, coexistent myocardial disease with cardiac dysfunction, found in 15–25% of cases with maternal autoantibody-induced AV block [93][9] and observed in left atrial isomerism, is associated with the development of FHF and fetal demise [92][8]. In mothers at high risk of having a child with CAVB, the use of HCQ may protect against recurrence of disease in a subsequent pregnancy.
2. Fetal Anemia
Fetal anemia either caused by alpha-thalassemia, isoimmunization or parvovirus B19 infection can be associated with hydrops fetalis. Fetal anemia may be a manifestation of twin-anemia-polycythemia sequence, seen in 3–5% of monochorionic twin pregnancies [94][10]. In Southeast Asia, fetal Hb Bart’s disease is the most common cause of hydrops fetalis, whereas isoimmunization and parvovirus B19 are commonly reported from the Western world. However, fetal anemia, in fact, rarely cause FHF, as believed in the past. Actually, a volume load secondary to increased cardiac output in fetuses with anemia can cause shifting of fluid from the vascular compartment to the interstitial space of the fetal body, in spite of the absence of CHF [23][11]. FHF secondary to fetal anemia developed only in the late stage of longstanding severe anemic hypoxia, usually long after the development of hydrops fetalis.
Of note, fetuses have high reserve potentials in responding to anemia. In spite of increased volume load, central venous pressure is not increased during the compensatory state, as indicated by increased forward flow in the ductus venosus during atrial contraction (markedly increased positive A-wave) (Figure 43) [19][12]. Several cases of fetuses with anemia show prolonged ICT [96][13], more pronounced than prolonged IRT as commonly seen in most cases developing FHF from other causes.
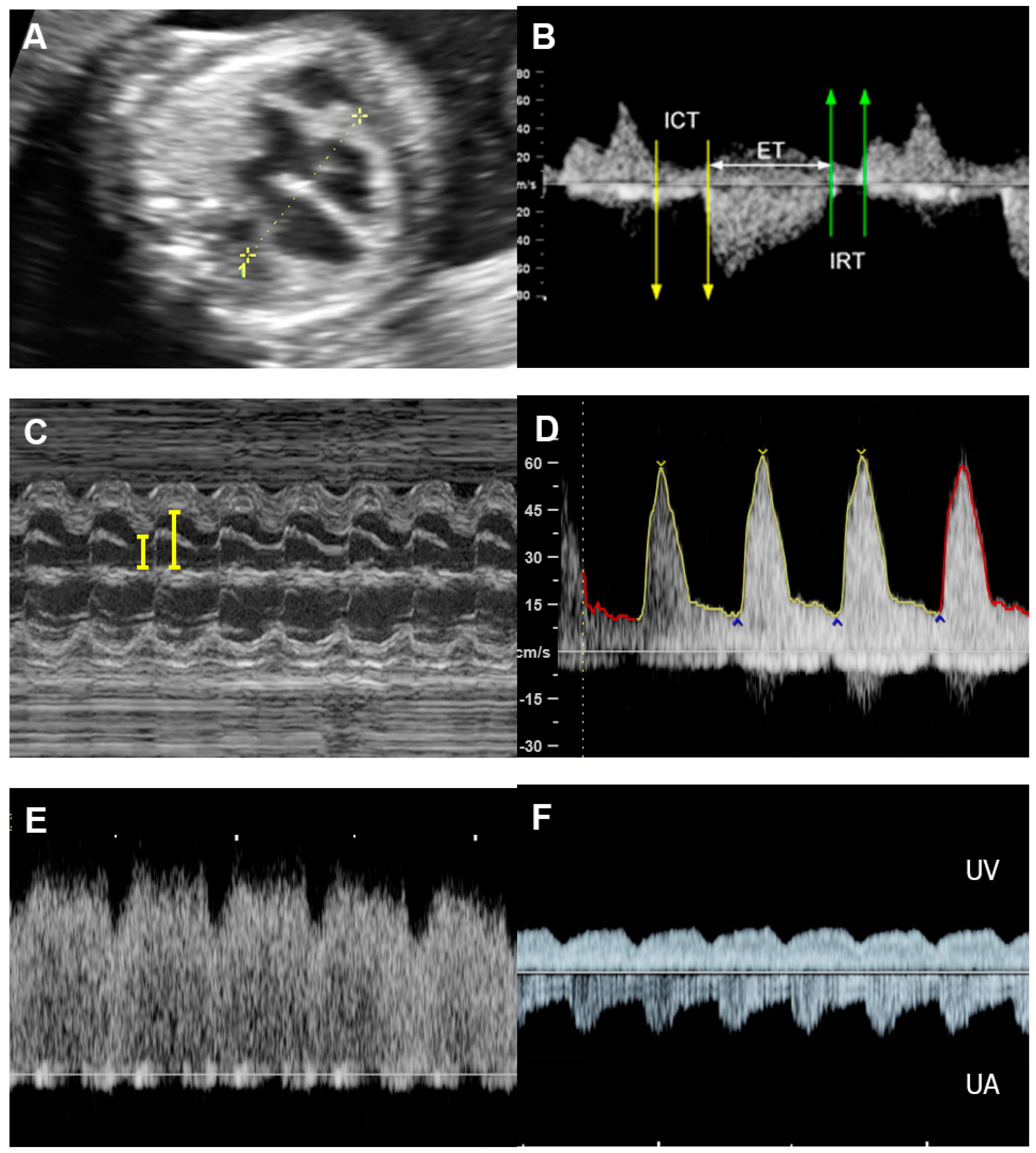
Figure 43. Hydrops without heart failure in Hb Bart’s disease: (A) cardiomegaly with effusions; (B) slightly increased Tei index (0.49) with prolonged ICT (42 ms); (C) normal shortening fraction 38%; (D) High MCA-PSV (60 cm/s); (E) markedly increased forward A-wave of the ductus venosus; (F) normal Doppler waveforms in the umbilical artery (UA) but pulsations in the umbilical vein (UV).
In fetal anemia, the oxygen-carrying capacity is reduced, leading to a decrease in systemic vascular resistance and an increase in cardiac output and widening of the arteriovenous O2 difference, as a compensatory mechanism [101,102][14][15]. Therefore, severe anemia can cause volume overload and increased stroke volume, which can alter ventricular function [103][16]. According to studies involving adaptive changes in post-transfusion anemic fetuses and studies on fetal lamb models, in fetuses with anemia, longitudinal strain, cardiac output, and shortening fraction are increased prior to intrauterine transfusion (IUT), when compared to the controls. Immediately after IUT, all parameters of systolic function are reduced to below the control levels, and 24 h after IUT, cardiac output, shortening fraction, and longitudinal strain are comparable with those of the controls [104,105,106][17][18][19]. This evidence suggests that a decrease in afterload is a primary mechanism of compensation in anemia, whereas an IUT increases intravascular volume and, therefore, flow resistance. A decrease in afterload caused by a reduction of serum viscosity may also play a role [103][16]. Additionally, anemia may cause peripheral vasodilation [107][20] associated with endothelial dysfunction [108][21]. In some fetuses with longstanding anemia, ventricular hypertrophy has also been detected in association with elevated catecholamines released in response to anemic hypoxia [109][22]. Nevertheless, ventricular hypertrophy is relatively rare in fetuses with anemia. Of note, cardiac compromise is more often seen in anemia caused by parvovirus-B19 infection, likely related to coexistent myocarditis [110][23]. Additionally, reduced colloid oncotic pressure, hypoxia-induced capillary damage and, possibly, portal hypertension can contribute to hydrops development [19][12]. As mentioned earlier, anemia-associated hydrops fetalis is not a sign of FHF in most cases.
Treatment of fetal anemia depends on the underlying causes. For example, termination of pregnancy is usually offered for lethal conditions such as fetal Hb Bart’s disease, whereas IUT is recommended in most cases of severe anemia secondary to parvovirus B19, Rh isoimmunization or Hb H hydrops fetalis. Current guidelines recommend cordocentesis to measure fetal Hb in case of MCA-PSV greater than 1.5 MoM and IUT if fetal hematocrit is less than 30% [111][24].
3. Non-Anemic High Cardiac Output
Several disorders are associated with an increase in combined cardiac output, such as arteriovenous malformations, the recipient of twin-to-twin transfusion syndrome and twin reversed arterial perfusion (TRAP) sequence (Figure 54), in which an acardiac twin without a heart or with a rudimentary heart is perfused by a pump twin [26][25]. Arteriovenous malformations most commonly occur in a highly vascularized mass, such as a placental chorioangioma, sacrococcygeal teratoma (Figure 65), hepatic hemangioma and aneurysm of vein of Galen. In these conditions, left to right shunting in the lesions causes systemic volume overload and is associated with increasing ventricular and atrial filling pressures.
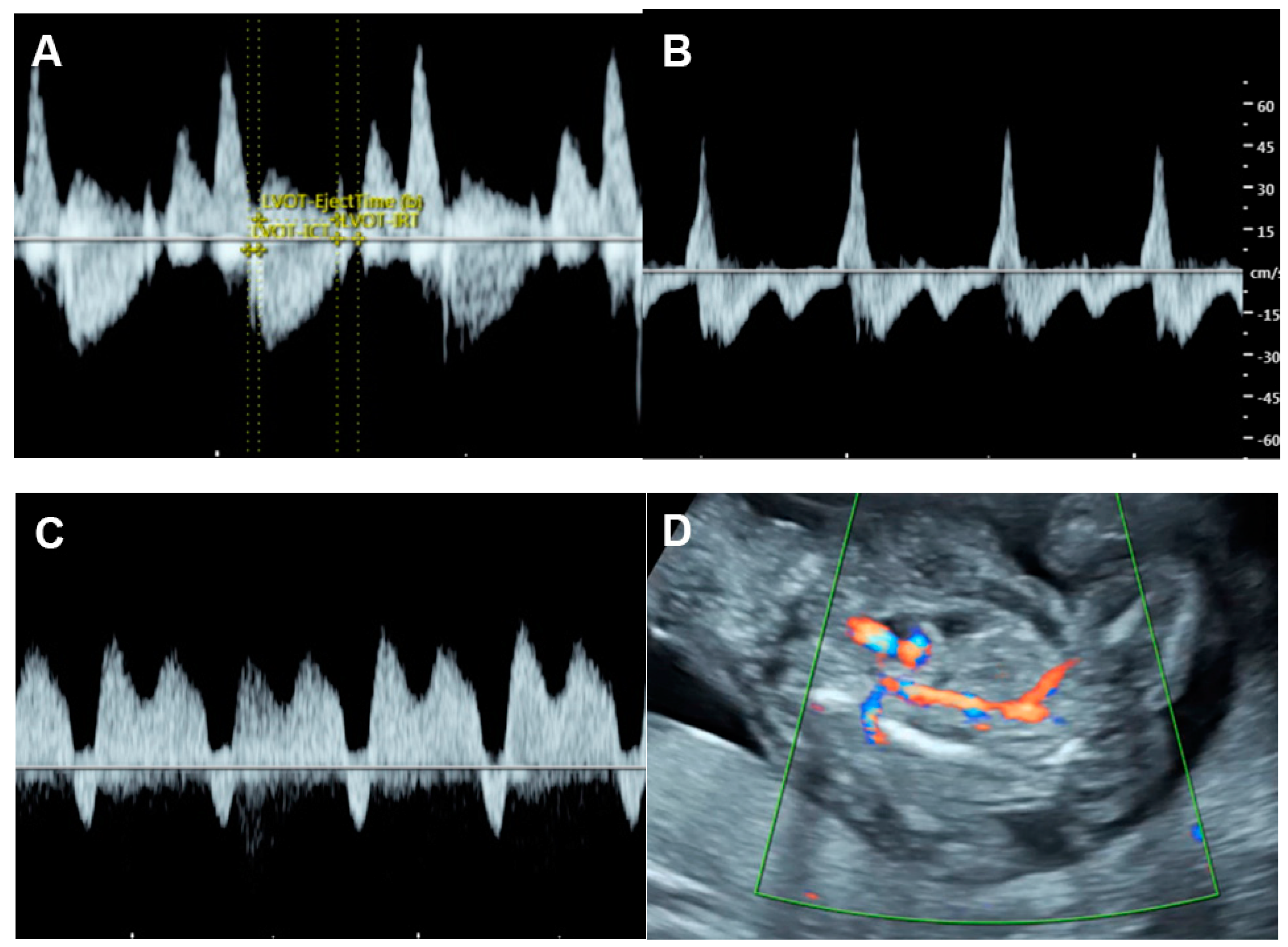
Figure 54. Cardiac dysfunction of the pumping twin in twin reversed arterial perfusion sequence: (A) increased Tei index (0.61) with prolonged IRT (46 ms); (B) Doppler flow in IVC; markedly re-versed A-wave (45 cm/s); (C) reversed A-wave in the ductus venosus; (D) acardiac twin with vas-cularization.
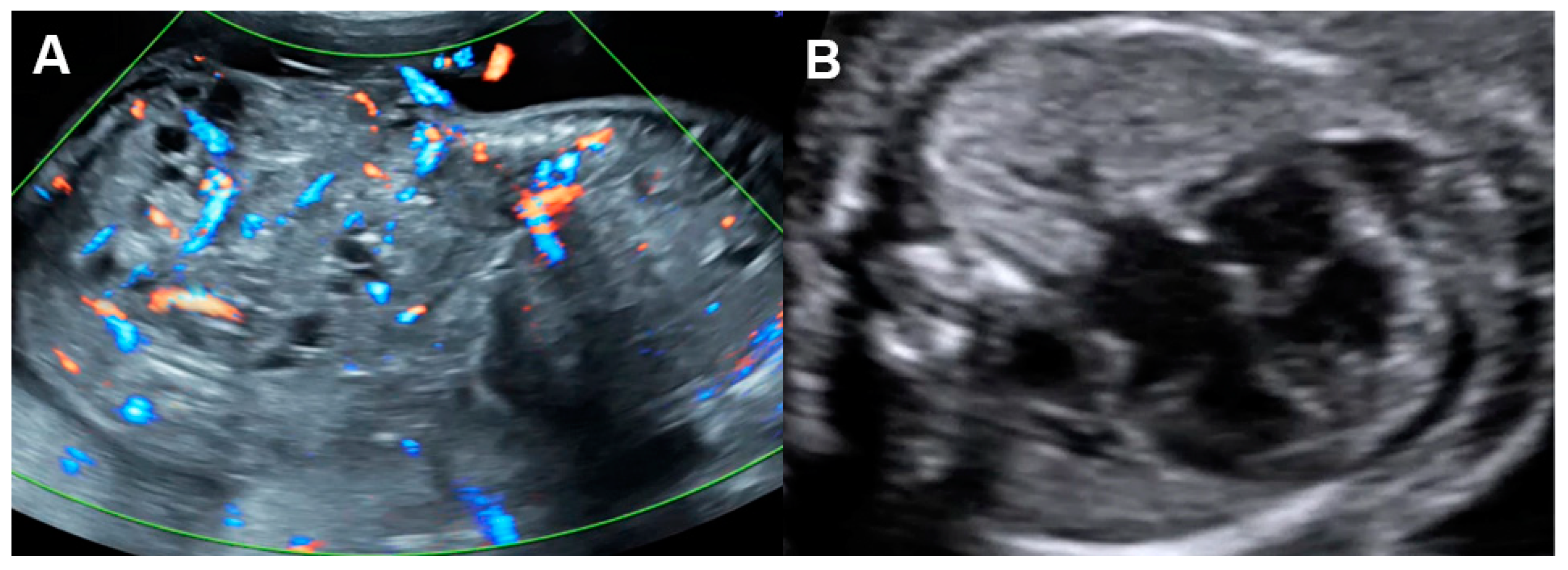
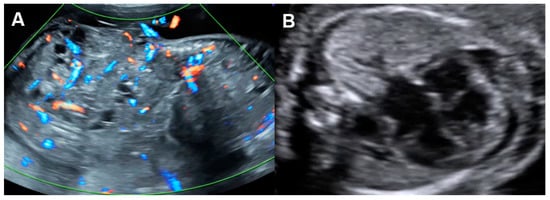
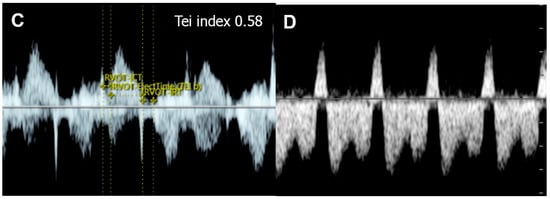
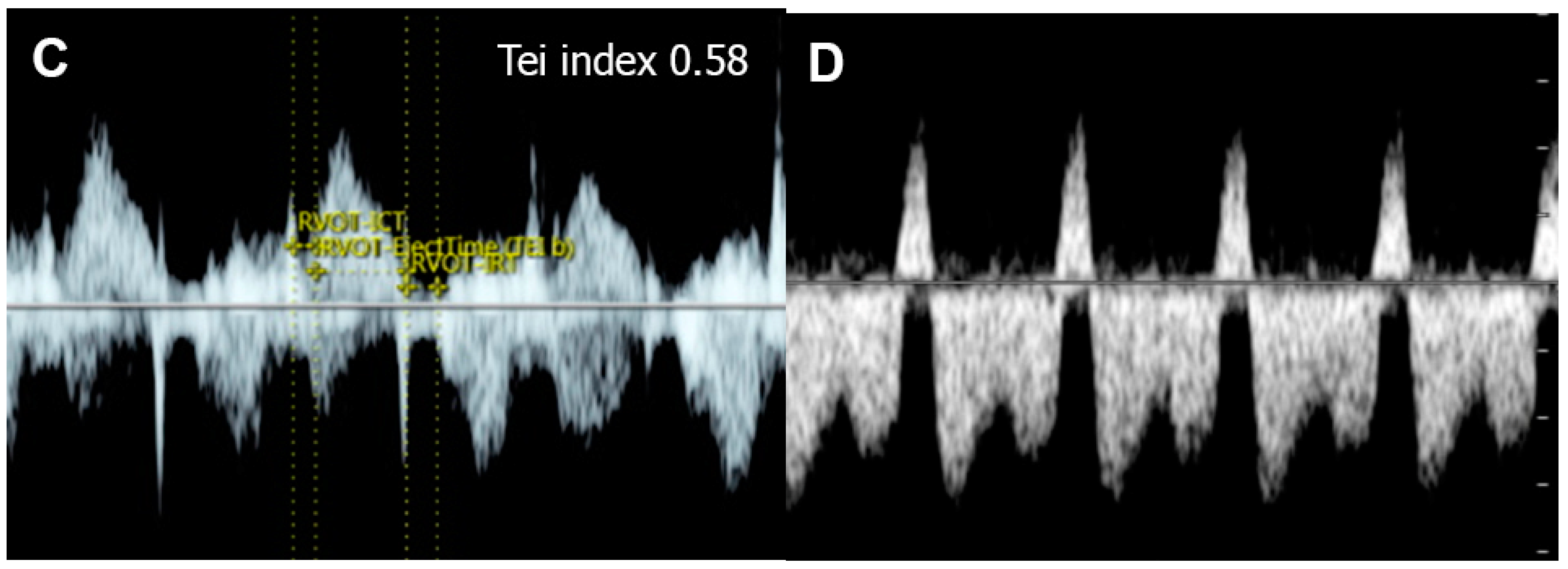
Figure 65. Cardiac dysfunction of a fetus with sacrococcygeal teratoma (SCT): (A) vascularized SCT; (B) global cardiomegaly with pericardial effusion; (C) increased Tei index (0.58) with prolonged IRT (44 ms); (D) Doppler markedly reversed A-wave in the ductus venosus.
The severity of cardiac overload is directly related to the volume of shunting, usually directly correlated with the tumor size or the acardiac twin size relative to fetal weight [112,113][26][27]. The cases with higher shunting volume or huge tumor mass have a high perinatal morbidity and mortality rate. As consequences of volume overload, the fetuses develop global cardiomegaly, increased cardiac output and cardiac dysfunction in varying degrees of severity. Typically, to cope with the cardiac overworking, compensatory afterload reduction occurs. However, when compensatory afterload reduction is overwhelmed, AV valve insufficiency or low CVPs can be expected. The measurement of combined cardiac output may be used to predict fetal compromise [114][28] and to guide the need for intrauterine intervention. It has been suggested that combined cardiac outputs of two-fold or greater of the reference range may be a cut-off for the development of FHF in sacrococcygeal teratoma or agenesis of the ductus venosus [115,116][29][30]. Fetal hydropic changes or fluid collection in the body cavities may develop in severe cases [117][31], though cardiac contractility or shortening fraction may be normal, hyperdynamic, or poor in advanced stages. Fetal echocardiography usually shows some degree of cardiac compromise, especially an increase in cardiac output [118,119][32][33] and a lower cardiovascular profile score (CVPs) [120][34], as well as abnormalities of venous Doppler waveforms. These fetal echocardiographic findings are also very helpful as guidance for prenatal intervention, such as obliteration or embolization of the main vessels feeding the tumor or perivascular sclerosis, surgical debulking of the tumor, arterial ligation, radiofrequency ablation of vessels of an acardiac twin, etc. Additionally, serial assessment of cardiac function in less severe cases is necessary for early detection of cardiac dysfunction or decompensation. The useful parameters for assessment include cardiac size, ventricular dilatation, combined cardiac output, spectral Doppler flow of the IVC, the ductus venosus or the umbilical vein as well as severity of AV valve regurgitation. Of note, different from volume load caused by anemia in which low viscosity and a marked decrease in afterload play an important role in the reduction of cardiac pressure load, the volume load in fetuses with non-anemic high output is associated with more rapid and severe cardiac pressure load and is more likely to cause FHF than anemic causes.
Increased Afterload
Increased ventricular afterload can eventually result in FHF, when compensatory mechanisms become exhausted. The causes of increased afterload are as follows: (1) blood flow obstruction in the heart, such as aortic stenosis, pulmonary stenosis, restrictive foramen ovale and constriction of the ductus arteriosus. (2) Extra-cardiac causes, including increased systemic vascular resistance caused by placental insufficiency as commonly seen in fetal growth restriction and twin-twin transfusion syndrome (TTTS).
4. Twin-to-Twin Transfusion Syndrome (TTTS)
TTTS is one of the most recognized causes of elevated afterload in the recipient twin. TTTS occurs in approximately 15–20% of monochorionic twin pregnancies. Pathologically, TTTS is caused by imbalance of blood flow through placental vascular anastomoses between the twins, with umbilical/chorionic arteries of the donor perfusing deep into the cotyledons of the recipient. In TTTS, the twins develop discrepancies in growth, blood volume, amniotic fluid volume and cardiac size. While the donor is typically small-for-date with oligohydramnios, the recipient suffers from volume overload. Because of hemodynamic imbalance, the donor has hypovolemia, while the recipient has volume overload, leading to a discrepancy in the fetal hemodynamic responses of both twins. The recipient twin shows an increase in afterload in response to volume load by releasing vasoactive peptides, probably originating from the placenta. The blood levels of several vasoconstrictors, such as endothelin-1, renin, and angiotensin- II, are markedly increased in the recipient twin [121][35], contributing to an increase in afterload. However, the donor twin may have high systemic vascular and placental resistance because of a compensatory response to a decrease in intravascular volume, but less alteration in cardiac function is often observed.
On fetal echocardiography, the recipient twin typically shows enlarged cardiac size; progressive biventricular hypertrophy, more pronounced on the right ventricle; elevated systemic pressures, leading to significant tricuspid and mitral regurgitation [122][36]; diastolic dysfunction with shortened filling fraction time; monophasic inflow of the E-A waveforms; increased Tei index primarily due to prolonged IRT [123][37]; and abnormal Doppler flow in the ductus venosus and IVC. Both diastolic and systolic dysfunction tend to be progressive and eventually result in FHF. The recipient twin has decreased ventricular strains of both sides [124[38][39],125], whereas the donor twin often shows a reduced strain rate of the right ventricle [125][39] but an increased left ventricular strain rate, likely caused by lower cerebrovascular resistance; however, placental resistance is increased. Differential circulation in the donor contributes to an increase in brain diastolic blood flow or cephalization, previously known as brain sparing, also associated with the response of myocardial deformation to alterations in differential afterload [125][39].
5. Fetal Growth Restriction (FGR)
Normally structural fetuses with growth restriction are commonly caused by uteroplacental insufficiency (UPI) which is closely associated with increased placental resistance, resulting in reduced end-diastolic Doppler velocity in the umbilical artery and increased cardiac afterload.
UPI results in chronic fetal hypoxia, which affects cardiac function with various mechanisms in FGR. In the early onset, FGR adaptive mechanisms involve the diversion of the cardiac output preferentially in favor of the brain (cephalization) and the heart. In the case of further worsening of fetal hypoxia, in addition to abnormal arterial flow, cardiac dysfunction and abnormal venous flow eventually occurs in order. In the uterine artery ligation rat model of FGR, growth-restricted fetal hearts had reduced wall thickness-to-diameter ratio, indicating LV dilatation, and they had elevated E/A ratios and a reduced ventricular shortening fraction, suggesting systolic and diastolic dysfunction, similar to human FGR [126][40].
In UPI-associated FGR (Figure 76), placental resistance is increased, indicated by an increase in systolic/diastolic ratio or pulsatility index of the umbilical artery or AEDV/REDV in severe cases. Increased placental resistance results in cardiac afterload, leading to redistribution of fetal hemodynamics. Because blood flow to the placenta, mostly running through the right ventricle, pulmonary artery and ductus arteriosus, is more difficult due to high resistance, blood circulation in the right atrium preferably crosses the foramen ovale, passing through the left atrium, the left ventricle, and the aorta to perfuse the brain, leading to an increase in cerebral blood flow, known as cephalization or brain-sparing effect. End-diastolic flow in the middle cerebral artery (MCA) is increased, resulting in a decreased MCA-PI/UA-PI ratio. In FGR, because of longstanding exposure to high cardiac afterload, right ventricular hypertrophy and cardiac dysfunction subsequently occur. Typically, when the compensatory mechanism coping with high afterload becomes overwhelmed, an abnormally high Tei index, poor shortening fraction and low cardiac output occur, followed by an increase in systemic venous pressure or increased preload index in the ductus venosus and IVC, represented by abnormally pronounced A-waves. Finally, when cardiac decompensation is more obvious, cardiac Doppler pulsation signals are strong enough to transmit through the ductus venosus to the umbilical vein, leading to the development of umbilical vein pulsations, which is commonly seen in the late phase of cardiac decompensation.
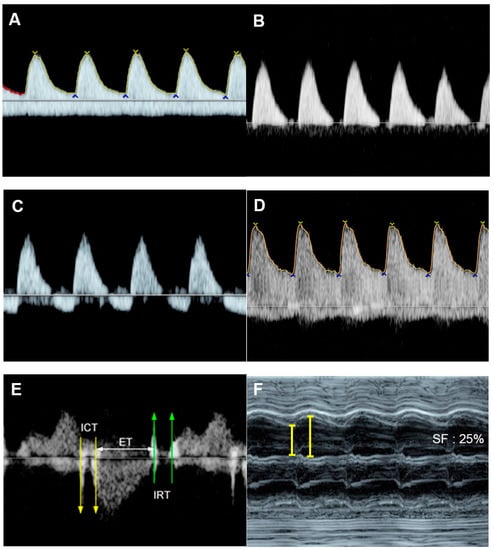
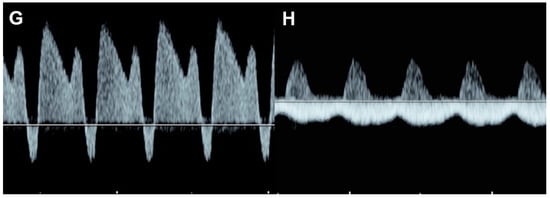
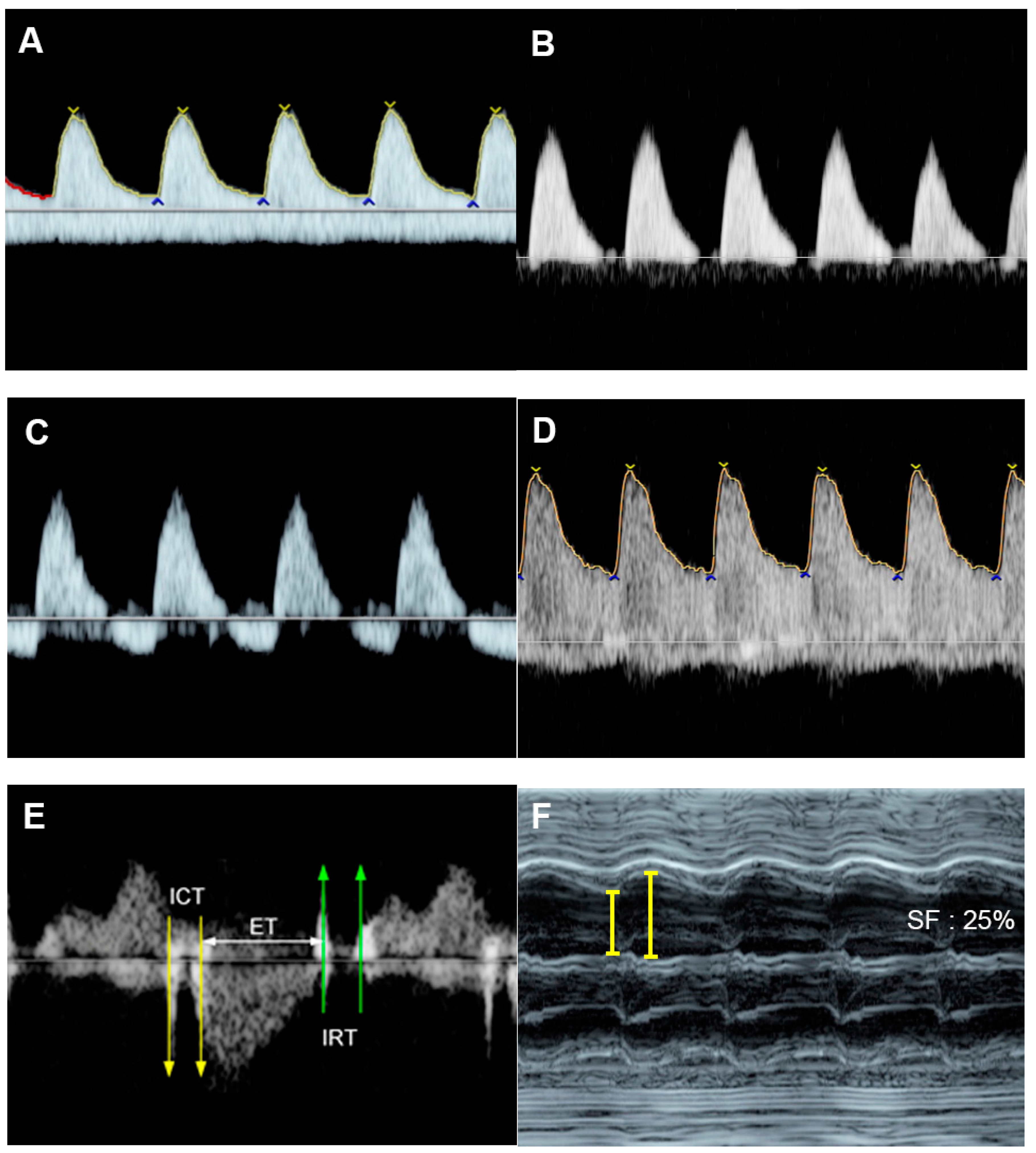
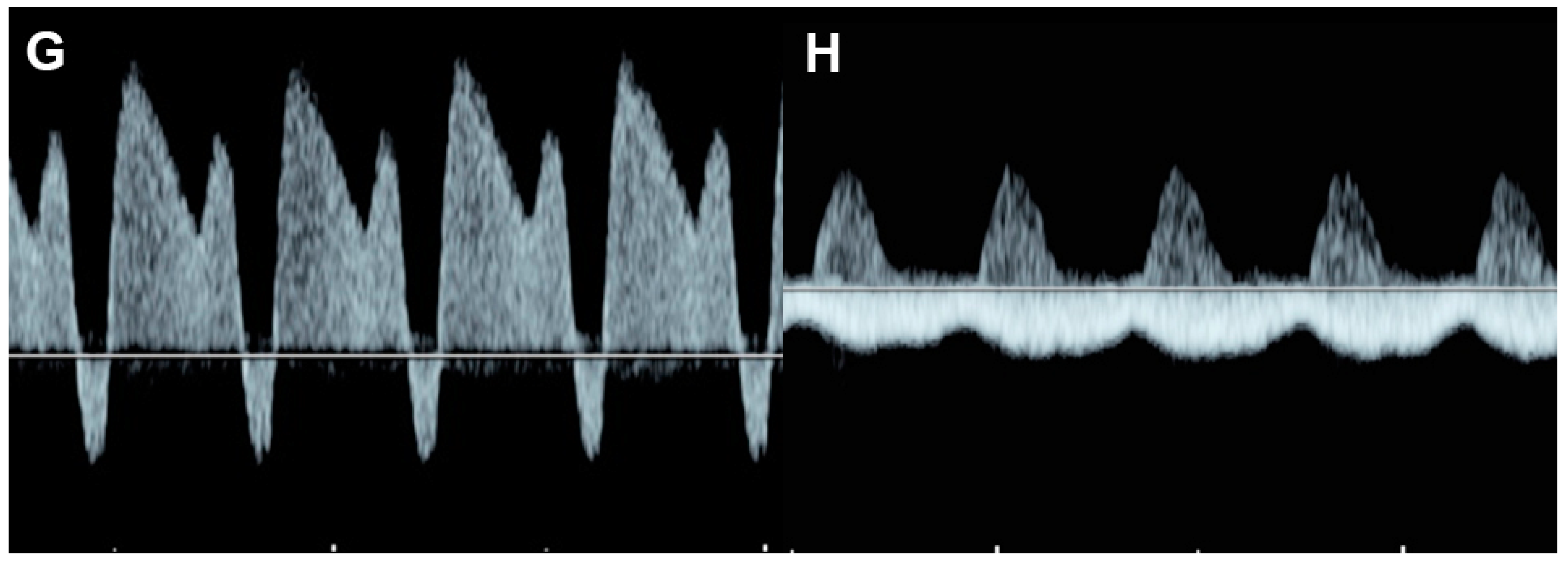
Figure 76. Fetal growth restriction: (A–C) Progressive changes of Doppler waveforms of the umbilical artery from low-end-diastolic flow at 27 weeks, to absent end-diastolic flow (B), and reversed end-diastolic flow (C) at 32 weeks; (D) Increased end-diastolic flow in the middle-cerebral artery (cephalization); (E) increased Tei index (0.61) with prolonged IRT (0.52 ms); (F) shortening fraction of 25%. (G) reversed A-wave in the ductus arteriosus; (H) pulsations in the umbilical vein at 32 weeks.
In response to chronic increased afterload, ventricular hypertrophy is commonly present in FGR [127][41]. Additionally, nearly half of the fetuses show sonographic signs of cardiac dysfunction. Additionally, cardiomegaly, monophasic inflow of the EA waveforms and holosystolic tricuspid regurgitation have been shown to be associated with adverse outcomes [74][42]. A study about cardiac function in FGR, assessed by cardio-STIC, demonstrated that reduced stroke volume occurred at the initial stage of fetal deterioration before the development of abnormal EF in FGR fetuses [128][43]. The authors suggest that stroke volume could be a sensitive indicator of cardiac dysfunction and is possibly better than EF measurement. In twin pregnancy, selective intrauterine growth restriction may occur in one twin in the absence of any other sonographic features of TTTS. As seen in FGR of singleton pregnancy, ventricular hypertrophy with dysfunction may develop and progress to FHF or even hydrops fetalis [129][44].
6. Critical Aortic Stenosis
Aortic stenosis (Figure 87) causes increased left ventricular afterload (leading to left ventricular systolic and diastolic dysfunction), ventricular growth failure (ending up with poor function or non-function) or left ventricular failure and development of endocardial fibroelastosis.
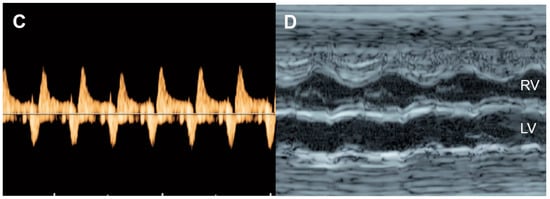
Figure 87. Critical aortic stenosis: (A,B) four-chamber views show enlarged LV with echogenic endocardium (fibroelastosis), mitral regurgitation, reversed flow across the foramen ovale; (C) markedly increased reversed A-wave in the pulmonary vein, indicating high pressure; (D) M-mode shows markedly poor contractility of the LV, compared to RV.
On echocardiography, typical features of critical aortic stenosis are as follows: an abnormal four-chamber view with left ventricular dilation, poor contractility or systolic dysfunction of the dilated left ventricle. The apex may still be formed by the dilated left ventricle, and the left atrium may be dilated because of mitral valve regurgitation. Pulsed Doppler shows forward flow across patent but stenotic aortic valve with high peak systolic velocity (generally greater than 200 cm/s), reversed flow in the aortic arch, reversed flow crossing the foramen ovale and monophasic and shortened mitral valve inflow time, indicating poor compliance of the ventricular wall and increased end-systolic pressure [130][45]. Reduction in aortic peak systolic velocity on follow-up ultrasound is a sign of progressive dysfunction of the left ventricle.
Bright echogenicity of the endocardium, reflecting endocardial fibroelastosis, is commonly seen in critical aortic stenosis and is associated with failure of biventricular repair [131][46].
Intrauterine intervention to relieve the aortic valve obstruction can improve both systolic and diastolic function and increase ejection fraction and mitral valve inflow time [132][47]. Additionally, RV-to-LV length ratio combined with LV pressure estimates, prior to fetal aortic valvuloplasty, may be used to predict a successful biventricular outcome with high sensitivity and specificity [133][48].
7. Ductal Arteriosus Constriction
Ductus arteriosus constriction (DAC) is most commonly associated with the use of nonsteroidal anti-inflammatory medications, especially indomethacin, in the late middle to early third trimester [38][49]. The constriction can cause an acute increase in right ventricular afterload, culminating in heart failure. Nevertheless, in cases of indomethacin-induced constriction, stopping such a therapy can reverse the constriction of the ductus within days.
On fetal echocardiography, the four-chamber view usually shows right ventricular hypertrophy and dilatation with impaired systolic function in many cases. Tricuspid regurgitation and right atrium enlargement is commonly visualized, reflecting elevated right ventricular systolic pressure. In acute cases, poor contractility of the right ventricle or ejection fraction may be demonstrated, likely caused by acute afterload elevation without compensatory myocardial hypertrophy. Pulsed Doppler shows high peak systolic velocities in the constricted ductus arteriosus, approximately 200–300 cm/s, as well as high diastolic velocities with a ductal pulsatility index of less than 1.9 [134][50]. The Tei index of the right ventricle is increased, associated with shortened ejection time, and returns to baseline coincident with ductal relaxation [135][51]. Monophasic inflow waveforms of the right ventricle are commonly seen. Tricuspid regurgitation and right atrium enlargement can predict the immediate prognosis of the newborn and provide guidance for the clinical judgment of the timing of pregnancy termination [136][52].
7.1. Intrinsic Contractile Dysfunction
Cardiomyopathy refers to a wide range of genetic, metabolic, familial, and inflammatory myocardial disorders in the absence of a structural cardiac defect in most cases. It is a disease of the myocardium affecting the LV, RV, or both and is commonly associated with abnormal cardiac function. It is relatively rare, accounting for 2.5% of fetal heart disease [137][53]. Based on echocardiography, cardiomyopathy is typically classified into two phenotypes; hypertrophic and dilated cardiomyopathy. The etiologies of both phenotypes are idiopathic in most cases (50%), whereas some are associated with genetic and inflammatory diseases. The most well-known disorder found in association with a hypertrophic cardiomyopathy is maternal diabetes mellitus. Dilated/non-hypertrophic cardiomyopathy, representing two-thirds of cases, is generally recognized as an enlarged heart with a dilated left ventricle, right ventricle, or commonly both ventricles. Echocardiography shows a decrease in left or right ventricular shortening fraction in most cases and a normal septal thickness. Ventricular dilation is demonstrated in less than 25% of cases. Color or pulsed Doppler shows some degree of regurgitation of the valve of the affected ventricle.
Hypertrophic cardiomyopathy (Figure 98), accounting for one-third of cases, is generally recognized as an enlarged heart in association with ventricular wall hypertrophy of one or, generally, both ventricles. Echocardiographic features are cardiomegaly or increased cardiothoracic ratio (80% of cases) with increased ventricular myocardial thickness, defined as a Z-score of greater than 2, and a normal left and right ventricular end-diastolic diameter in the absence of anatomical or systemic diseases.
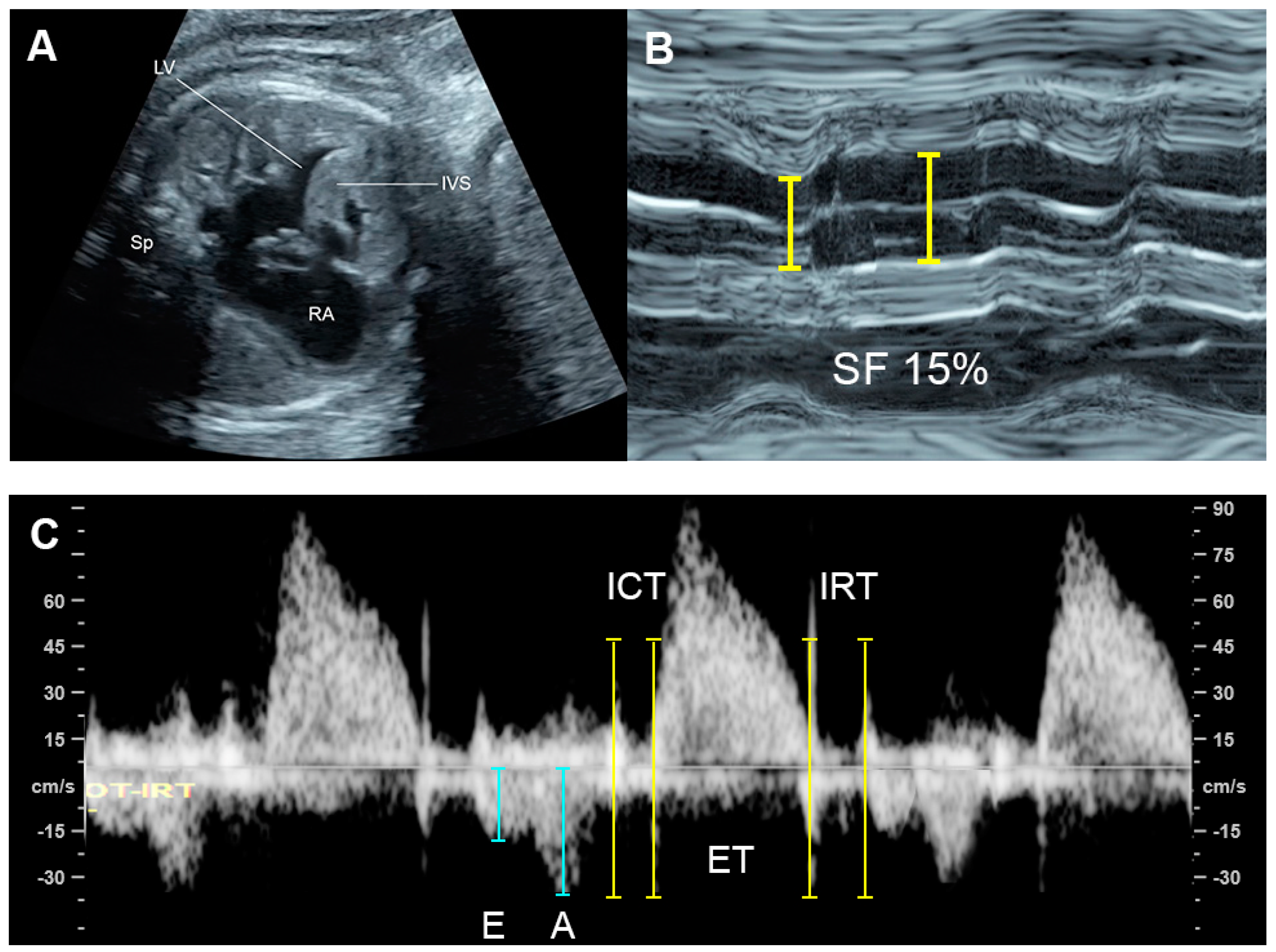
Figure 98. Hypertrophic cardiomyopathy: (A) four-chamber view: thickened ventricular wall and interventricular septum; (B) M-mode: poor contractility with a shortening fraction of 15%; (C) Car-diac PW Doppler: Low E/A ratio, prolonged IRT and increased Tei index (ICT: 40 ms; ET 154 ms; IRT 54 ms).
The prognosis of cardiomyopathy depends on the underlying associated abnormalities. Generally, a poor prognosis may be expected in cases of poor diastolic dysfunction such as monophasic inflow, umbilical vein pulsations, and hydrops fetalis [93,137][9][53]. Non-hypertrophic cardiomyopathy appears to have a better prognosis than hypertrophic cardiomyopathy. However, ventricular hypertrophy associated with maternal diabetes has a better prognosis [93][9].
7.2. Congenital Heart Defect
Though congenital heart defects (CHD) are common, most of them are not associated with FHF. Approximately 5% of CHD are associated with heart failure [138][54] or CVPs of 7 or less [72][55]. Common CHD associated with cardiac failure are as follows [139][56]:
-
Critical aortic stenosis
-
Ebstein anomaly
-
Severe atrioventricular valve insufficiency
-
Severe semilunar valve insufficiency
-
Tetralogy of Fallot with absent pulmonary valves
-
Pulmonary atresia with severe tricuspid valve insufficiency
-
Truncus arteriosus with severe incompetent truncal valves
-
Bilateral outflow tract obstruction
-
Intracardiac tumors
-
Single ventricle physiology with ventricular dysfunction
The majority of fetuses with CHD are well tolerated. This is because most cases have at least one well-functioning ventricle, contributing to redistribution of the systemic and pulmonary venous blood flow through the normal ventricle which can maintain the equivalent of a combined cardiac output without an increase in central venous and atrial filling pressures. Gembruch et al. [140][57] demonstrated that venous Doppler in fetuses with isolated CHD did not present sufficient alterations to be a reliable marker for screening purposes for CHD in second- and third-trimester fetuses. Abnormal venous Doppler results were mainly attributable to myocardial dysfunction and also to severe right heart obstruction even in the absence of congestive heart failure. Therefore, venous Doppler studies are clinically helpful in indirectly monitoring cardiac function in fetuses with CHD. When abnormal central venous flow patterns do occur in association with cardiac defects in utero, they are usually caused by other processes occurring simultaneously that affect ventricular compliance (e.g., endocardial fibroelastosis) or rhythm-related hemodynamics (e.g., complete heart block) [141][58].
CHD with dysfunction of both ventricles or with biventricular inflow or outflow obstruction, venous and atrial pressure load can occur and result in FHF. CHD associated with markedly increased afterload such as critical aortic stenosis, acute constriction of ductus arteriosus, truncus arteriosus with truncal valve stenosis are usually poorly tolerated, likely leading to abnormal ventricular dysfunction or FHF. CHD with volume load as commonly seen in atrioventricular and semilunar valve insufficiency are also likely to evolve FHF. For example, the Ebstein anomaly is usually related to severe tricuspid dysfunction and volume load (Figure 109), which eventually compromises left ventricular filling as well as global function and combined cardiac output, leading to hydrops fetalis [142][59]. Likewise, Inamura et al. [143][60] demonstrated that global left ventricular performance as assessed by the Tei index was abnormal in severe tricuspid valve dysplasia and likely to develop hydrops or result in mortality. Probably, markedly enlarged right-sided heart causes a mechanical compression of the left ventricle, leading to limitation of left ventricle filling. Additionally, in case of no antegrade flow through the right outflow tract and pulmonary trunk, total venous preload is redistributed to the left heart, resulting in left ventricular dysfunction secondary to excessive volume load and inability to maintain the equivalent of a combined cardiac output, ultimately evolving to FHF.
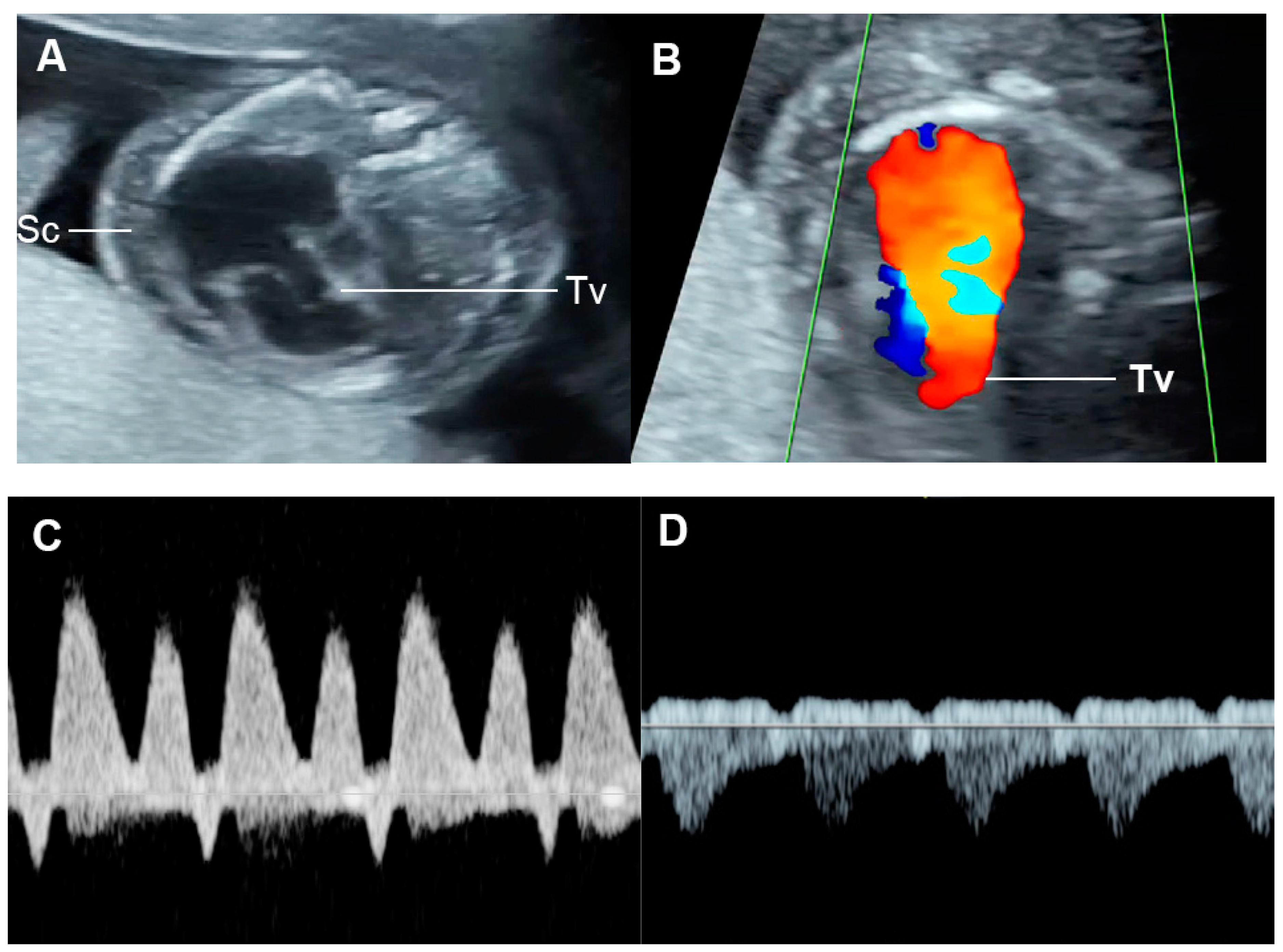
Figure 109. Ebstein anomaly with hydrops fetalis: (A) four-chamber view: cardiomegaly, low inser-tion of septal leaflet of the tricuspid valve (Tv) and subcutaneous edema (Sc); (B) color flow mapping of the four-chamber view: severe tricuspid regurgitation with originating point near the apex; (C) ductus venosus Doppler: reversed A-wave; (D) venous pulsations of the umbilical vein.
7.3. External Cardiac Compression
External cardiac compression of the fetal heart can occur in cases of congenital diaphragmatic hernia (CDH) [144][61], congenital cystic adenomatoid malformation (Figure 110) [145][62], tumors such as mediastinal or pericardial teratoma [146,147][63][64] and massive pleural effusions [148][65]. These conditions can compress the heart or distort and compress the systemic veins returning to the heart, leading to reduced cardiac size and ventricular filling, resulting in low cardiac output and increased central venous pressures and eventually FHF or hydrops fetalis. Such lesions in some cases can also more directly influence lymphatic drainage and may be a primary cause of evolving fetal hydrops fetalis [149][66]. Fetal echocardiography usually shows diastolic dysfunction (increased Doppler E/A ratio and abnormal venous Doppler) together with reduced cardiac size (cardio-thoracic area ratio).
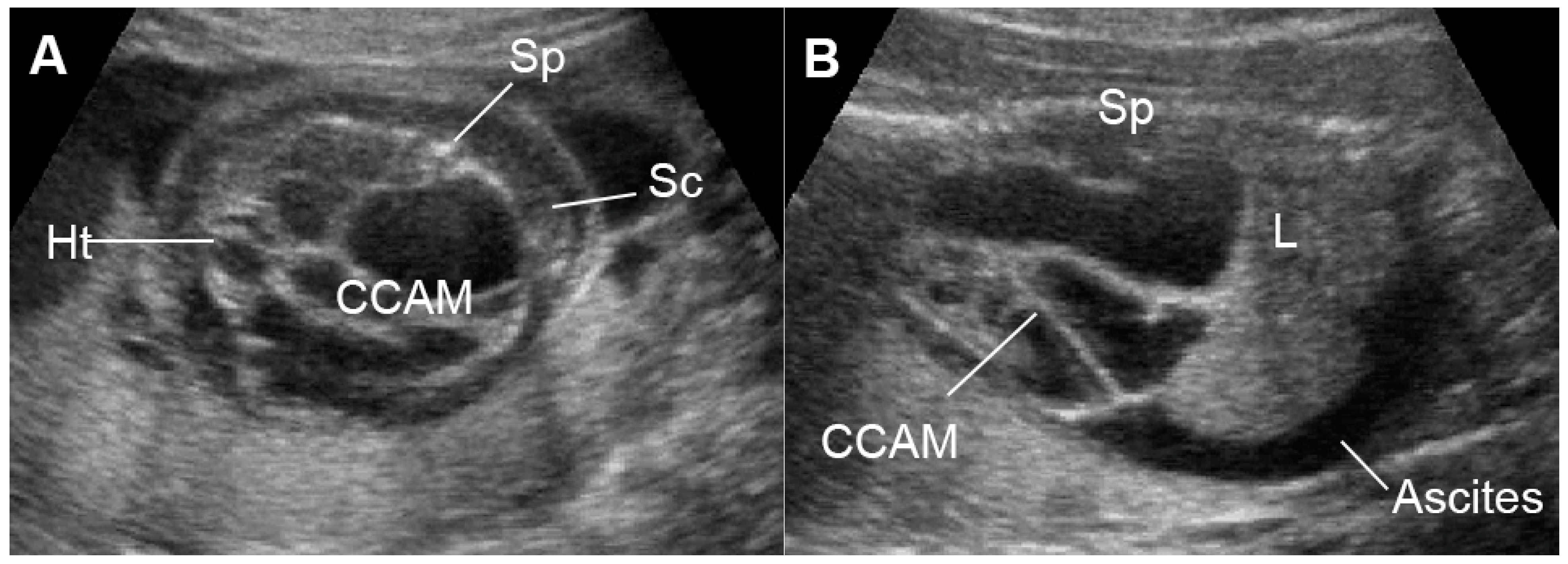
Figure 110. Large cystic adenomatoid malformation (CCAM) type I with hydrops fetalis: (A) cross-sectional scan at the four-chamber view: small heart compressed by the cyst and marked subcutaneous edema (Sc); (B) sagittal scan of the fetal trunk: CCAM occupying the entire chest and ascites (L: liver; Sp: spine).
References
- Hornberger, L.K.; Sahn, D.J. Rhythm abnormalities of the fetus. Heart 2007, 93, 1294–1300.
- Demirci, O.; Tosun, Ö.; Bolat, G. Prenatal diagnosis and management of fetal supraventricular tachyarrhythmia and postnatal outcomes. J. Gynecol. Obstet. Hum. Reprod. 2022, 51, 102323.
- Yuan, S.M. Fetal arrhythmias: Diagnosis and treatment. J. Matern. Fetal Neonatal Med. 2020, 33, 2671–2678.
- Shin, J.A.; Choi, Y.U.; Kim, K.M.; Yoon, J.H.; Lee, J.Y. Congenital Long QT Syndrome Type 2 with Symptomatic 2:1 Atrioventricular Block and Ventricular Arrhythmia in a Preterm Baby Who Presented with Fetal Ventricular Tachycardia and Hydrops. Korean Circ. J. 2021, 51, 792–796.
- Gembruch, U.; Krapp, M.; Baumann, P. Changes of venous blood flow velocity waveforms in fetuses with supraventricular tachycardia. Ultrasound Obstet. Gynecol. 1995, 5, 394–399.
- Schmidt, K.G.; Ulmer, H.E.; Silverman, N.H.; Kleinman, C.S.; Copel, J.A. Perinatal outcome of fetal complete atrioventricular block: A multicenter experience. J. Am. Coll. Cardiol. 1991, 17, 1360–1366.
- Jaeggi, E.T.; Hornberger, L.K.; Smallhorn, J.F.; Fouron, J.C. Prenatal diagnosis of complete atrioventricular block associated with structural heart disease: Combined experience of two tertiary care centers and review of the literature. Ultrasound Obstet. Gynecol. 2005, 26, 16–21.
- Berg, C.; Geipel, A.; Kohl, T.; Breuer, J.; Germer, U.; Krapp, M.; Baschat, A.A.; Hansmann, M.; Gembruch, U. Atrioventricular block detected in fetal life: Associated anomalies and potential prognostic markers. Ultrasound Obstet. Gynecol. 2005, 26, 4–15.
- Pedra, S.R.; Smallhorn, J.F.; Ryan, G.; Chitayat, D.; Taylor, G.P.; Khan, R.; Abdolell, M.; Hornberger, L.K. Fetal cardiomyopathies: Pathogenic mechanisms, hemodynamic findings, and clinical outcome. Circulation 2002, 106, 585–591.
- Slaghekke, F.; Kist, W.J.; Oepkes, D.; Middeldorp, J.M.; Klumper, F.J.; Vandenbussche, F.P.; Lopriore, E. TAPS and TOPS: Two distinct forms of feto-fetal transfusion in monochorionic twins. Z Geburtshilfe Neonatol 2009, 213, 248–254.
- Thammavong, K.; Luewan, S.; Jatavan, P.; Tongsong, T. Foetal haemodynamic response to anaemia. ESC Heart Fail. 2020, 7, 3473–3482.
- Tongsong, T.; Tongprasert, F.; Srisupundit, K.; Luewan, S. Venous Doppler studies in low-output and high-output hydrops fetalis. Am. J. Obstet. Gynecol. 2010, 203, 488.e1–488.e6.
- Tongprasert, F.; Srisupundit, K.; Luewan, S.; Traisrisilp, K.; Jatavan, P.; Tongsong, T. Fetal isovolumetric time intervals as a marker of abnormal cardiac function in fetal anemia from homozygous alpha thalassemia-1 disease. Prenat. Diagn. 2017, 37, 1028–1032.
- Davis, L.E.; Hohimer, A.R. Hemodynamics and organ blood flow in fetal sheep subjected to chronic anemia. Am. J. Physiol 1991, 261, R1542–R1548.
- Kilby, M.D.; Szwarc, R.; Benson, L.N.; Morrow, R.J. Left ventricular hemodynamics in anemic fetal lambs. J. Perinat. Med. 1998, 26, 5–12.
- Metivier, F.; Marchais, S.J.; Guerin, A.P.; Pannier, B.; London, G.M. Pathophysiology of anaemia: Focus on the heart and blood vessels. Nephrol. Dial. Transplant. 2000, 15 (Suppl. 3), 14–18.
- Michel, M.; Schmitz, R.; Kiesel, L.; Steinhard, J. Fetal.l myocardial peak systolic strain before and after intrauterine red blood cell transfusion—A tissue Doppler imaging study. J. Perinat. Med. 2012, 40, 545–550.
- Rizzo, G.; Nicolaides, K.H.; Arduini, D.; Campbell, S. Effects of intravascular fetal blood transfusion on fetal intracardiac Doppler velocity waveforms. Am. J. Obstet. Gynecol. 1990, 163, 1231–1238.
- Nassar, R.; Reedy, M.C.; Anderson, P.A. Developmental changes in the ultrastructure and sarcomere shortening of the isolated rabbit ventricular myocyte. Circ. Res. 1987, 61, 465–483.
- Mehta, P.A.; Dubrey, S.W. High output heart failure. Qjm 2009, 102, 235–241.
- Teixeira, R.S.; Terse-Ramos, R.; Ferreira, T.A.; Machado, V.R.; Perdiz, M.I.; Lyra, I.M.; Nascimento, V.L.; Boa-Sorte, N.; Andrade, B.B.; Ladeia, A.M. Associations between endothelial dysfunction and clinical and laboratory parameters in children and adolescents with sickle cell anemia. PLoS ONE 2017, 12, e0184076.
- Oberhoffer, R.; Grab, D.; Keckstein, J.; Högel, J.; Terinde, R.; Lang, D. Cardiac changes in fetuses secondary to immune hemolytic anemia and their relation to hemoglobin and catecholamine concentrations in fetal blood. Ultrasound Obstet. Gynecol. 1999, 13, 396–400.
- von Kaisenberg, C.S.; Jonat, W. Fetal parvovirus B19 infection. Ultrasound Obstet. Gynecol. 2001, 18, 280–288.
- Mari, G.; Norton, M.E.; Stone, J.; Berghella, V.; Sciscione, A.C.; Tate, D.; Schenone, M.H. Society for Maternal-Fetal Medicine (SMFM) Clinical Guideline #8: The fetus at risk for anemia—Diagnosis and management. Am. J. Obstet. Gynecol. 2015, 212, 697–710.
- Eckersley, L.; Hornberger, L.K. Cardiac function and dysfunction in the fetus. Echocardiography 2017, 34, 1776–1787.
- Coleman, A.; Kline-Fath, B.; Keswani, S.; Lim, F.Y. Prenatal solid tumor volume index: Novel prenatal predictor of adverse outcome in sacrococcygeal teratoma. J. Surg. Res. 2013, 184, 330–336.
- Akinkuotu, A.C.; Coleman, A.; Shue, E.; Sheikh, F.; Hirose, S.; Lim, F.Y.; Olutoye, O.O. Predictors of poor prognosis in prenatally diagnosed sacrococcygeal teratoma: A multiinstitutional review. J. Pediatr. Surg 2015, 50, 771–774.
- Statile, C.J.; Cnota, J.F.; Gomien, S.; Divanovic, A.; Crombleholme, T.; Michelfelder, E. Estimated cardiac output and cardiovascular profile score in fetuses with high cardiac output lesions. Ultrasound Obstet. Gynecol. 2013, 41, 54–58.
- Jaeggi, E.T.; Fouron, J.C.; Hornberger, L.K.; Proulx, F.; Oberhänsli, I.; Yoo, S.J.; Fermont, L. Agenesis of the ductus venosus that is associated with extrahepatic umbilical vein drainage: Prenatal features and clinical outcome. Am. J. Obstet. Gynecol. 2002, 187, 1031–1037.
- Wilson, R.D.; Hedrick, H.; Flake, A.W.; Johnson, M.P.; Bebbington, M.W.; Mann, S.; Rychik, J.; Liechty, K.; Adzick, N.S. Sacrococcygeal teratomas: Prenatal surveillance, g.growth and pregnancy outcome. Fetal Diagn. Ther. 2009, 25, 15–20.
- Bond, S.J.; Harrison, M.R.; Schmidt, K.G.; Silverman, N.H.; Flake, A.W.; Slotnick, R.N.; Anderson, R.L.; Warsof, S.L.; Dyson, D.C. Death due to high-output cardiac failure in fetal sacrococcygeal teratoma. J. Pediatr. Surg 1990, 25, 1287–1291.
- Byrne, F.A.; Lee, H.; Kipps, A.K.; Brook, M.M.; Moon-Grady, A.J. Echocardiographic risk stratification of fetuses with sacrococcygeal teratoma and twin-reversed arterial perfusion. Fetal Diagn. Ther. 2011, 30, 280–288.
- Rychik, J. Fetal cardiovascular physiology. Pediatr. Cardiol. 2004, 25, 201–209.
- Donofrio, M.T.; Moon-Grady, A.J.; Hornberger, L.K.; Copel, J.A.; Sklansky, M.S.; Abuhamad, A.; Cuneo, B.F.; Huhta, J.C.; Jonas, R.A.; Krishnan, A.; et al. Diagnosis and treatment of fetal cardiac disease: A scientific statement from the American Heart Association. Circulation 2014, 129, 2183–2242.
- Bajoria, R.; Sullivan, M.; Fisk, N.M. Endothelin concentrations in monochorionic twins with severe twin-twin transfusion syndrome. Hum. Reprod. 1999, 14, 1614–1618.
- Mahieu-Caputo, D.; Salomon, L.J.; Le Bidois, J.; Fermont, L.; Brunhes, A.; Jouvet, P.; Dumez, Y.; Dommergues, M. Fetal hypertension: An insight into the pathogenesis of the twin-twin transfusion syndrome. Prenat. Diagn. 2003, 23, 640–645.
- Rychik, J.; Tian, Z.; Bebbington, M.; Xu, F.; McCann, M.; Mann, S.; Wilson, R.D.; Johnson, M.P. The twin-twin transfusion syndrome: Spectrum of cardiovascular abnormality and development of a cardiovascular score to assess severity of disease. Am. J. Obstet. Gynecol. 2007, 197, 392.e1–392.e8.
- Van Mieghem, T.; Giusca, S.; DeKoninck, P.; Gucciardo, L.; Doné, E.; Hindryckx, A.; D’Hooge, J.; Deprest, J. Prospective assessment of fetal cardiac function with speckle tracking in healthy fetuses and recipient fetuses of twin-to-twin transfusion syndrome. J. Am. Soc. Echocardiogr. 2010, 23, 301–308.
- Rychik, J.; Zeng, S.; Bebbington, M.; Szwast, A.; Quartermain, M.; Natarajan, S.; Johnson, M.; Tian, Z. Speckle tracking-derived myocardial tissue deformation imaging in twin-twin transfusion syndrome: Differences in strain and strain rate between donor and recipient twins. Fetal Diagn. Ther. 2012, 32, 131–137.
- Dai, Y.; Zhao, D.; Chen, C.K.; Yap, C.H. Echocardiographic assessment of fetal cardiac function in the uterine artery ligation rat model of IUGR. Pediatr. Res. 2021, 90, 801–808.
- Veille, J.C.; Hanson, R.; Sivakoff, M.; Hoen, H.; Ben-Ami, M. Fetal cardiac size in normal, intrauterine growth retarded, and diabetic pregnancies. Am. J. Perinatol 1993, 10, 275–279.
- Mäkikallio, K.; Räsänen, J.; Mäkikallio, T.; Vuolteenaho, O.; Huhta, J.C. Human fetal cardiovascular profile score and neonatal outcome in intrauterine growth restriction. Ultrasound Obstet. Gynecol. 2008, 31, 48–54.
- Ziyu, T. Assessment of left ventricular function by spatio-temporal image correlation in fetuses with fetal growth restriction. Echocardiography 2022, 39, 1240–1244.
- Kondo, Y.; Hidaka, N.; Yumoto, Y.; Fukushima, K.; Tsukimori, K.; Wake, N. Cardiac hypertrophy of one fetus and selective growth restriction of the oTher. fetus in a monochorionic twin pregnancy. J. Obstet. Gynaecol. Res. 2010, 36, 401–404.
- Mäkikallio, K.; McElhinney, D.B.; Levine, J.C.; Marx, G.R.; Colan, S.D.; Marshall, A.C.; Lock, J.E.; Marcus, E.N.; Tworetzky, W. Fetal aortic valve stenosis and the evolution of hypoplastic left heart syndrome: Patient selection for fetal intervention. Circulation 2006, 113, 1401–1405.
- McElhinney, D.B.; Vogel, M.; Benson, C.B.; Marshall, A.C.; Wilkins-Haug, L.E.; Silva, V.; Tworetzky, W. Assessment of left ventricular endocardial fibroelastosis in fetuses with aortic stenosis and evolving hypoplastic left heart syndrome. Am. J. Cardiol. 2010, 106, 1792–1797.
- Selamet Tierney, E.S.; Wald, R.M.; McElhinney, D.B.; Marshall, A.C.; Benson, C.B.; Colan, S.D.; Marcus, E.N.; Marx, G.R.; Levine, J.C.; Wilkins-Haug, L.; et al. Changes in left heart hemodynamics after technically successful in-utero aortic valvuloplasty. Ultrasound Obstet. Gynecol. 2007, 30, 715–720.
- Tulzer, A.; Arzt, W.; Gitter, R.; Sames-Dolzer, E.; Kreuzer, M.; Mair, R.; Tulzer, G. Valvuloplasty in 103 fetuses with critical aortic stenosis: Outcome and new predictors for postnatal circulation. Ultrasound Obstet. Gynecol. 2022, 59, 633–641.
- Beaute, J.I.; Friedman, K.G. Indomethacin induced ductus arteriosus closure in midgestation fetus. Clin. Case Rep. 2018, 6, 506–508.
- Tulzer, G.; Gudmundsson, S.; Sharkey, A.M.; Wood, D.C.; Cohen, A.W.; Huhta, J.C. Doppler echocardiography of fetal ductus arteriosus constriction versus increased right ventricular output. J. Am. Coll. Cardiol. 1991, 18, 532–536.
- Mori, Y.; Rice, M.J.; McDonald, R.W.; Reller, M.D.; Wanitkun, S.; Harada, K.; Sahn, D.J. Evaluation of systolic and diastolic ventricular performance of the right ventricle in fetuses with ductal constriction using the Doppler Tei index. Am. J. Cardiol. 2001, 88, 1173–1178.
- Wen, J.; Guo, X.; Cai, S.; Xu, D.; Zhang, G.; Bai, X. Fetal Ductus Arteriosus Premature Constriction. Int. Heart J. 2022, 63, 722–728.
- Weber, R.; Kantor, P.; Chitayat, D.; Friedberg, M.K.; Golding, F.; Mertens, L.; Nield, L.E.; Ryan, G.; Seed, M.; Yoo, S.J.; et al. Spectrum and outcome of primary cardiomyopathies diagnosed during fetal life. JACC Heart Fail. 2014, 2, 403–411.
- Machin, G.A. Hydrops revisited: Literature review of 1,414 cases published in the 1980s. Am. J. Med. Genet. 1989, 34, 366–390.
- Wieczorek, A.; Hernandez-Robles, J.; Ewing, L.; Leshko, J.; Luther, S.; Huhta, J. Prediction of outcome of fetal congenital heart disease using a cardiovascular profile score. Ultrasound Obstet. Gynecol. 2008, 31, 284–288.
- Ojala, T.H.; Hornberger, L.K. Fetal heart failure. Front. Biosci. 2010, 2, 891–906.
- Gembruch, U.; Meise, C.; Germer, U.; Berg, C.; Geipel, A. Venous Doppler ultrasound in 146 fetuses with congenital heart disease. Ultrasound Obstet. Gynecol. 2003, 22, 345–350.
- Huhta, J.C. Guidelines for the evaluation of heart failure in the fetus with or without hydrops. Pediatr. Cardiol. 2004, 25, 274–286.
- Pavlova, M.; Fouron, J.C.; Drblik, S.P.; van Doesburg, N.H.; Bigras, J.L.; Smallhorn, J.; Harder, J.; Robertson, M. Factors affecting the prognosis of Ebstein’s anomaly during fetal life. Am. Heart J. 1998, 135, 1081–1085.
- Inamura, N.; Taketazu, M.; Smallhorn, J.F.; Hornberger, L.K. Left ventricular myocardial performance in the fetus with severe tricuspid valve disease and tricuspid insufficiency. Am. J. Perinatol. 2005, 22, 91–97.
- Soni, S.; Moldenhauer, J.S.; Rintoul, N.; Adzick, N.S.; Hedrick, H.L.; Khalek, N. Perinatal Outcomes in Fetuses Prenatally Diagnosed with Congenital Diaphragmatic Hernia and Concomitant Lung Lesions: A 10-Year Review. Fetal Diagn. Ther. 2020, 47, 630–635.
- Kane, S.C.; Da Silva Costa, F.; Crameri, J.A.; Reidy, K.L.; Kaganov, H.; Palma-Dias, R. Antenatal assessment and postnatal outcome of fetal echogenic lung lesions: A decade’s experience at a tertiary referral hospital. J. Matern. Fetal Neonatal Med. 2019, 32, 703–709.
- Rychik, J.; Khalek, N.; Gaynor, J.W.; Johnson, M.P.; Adzick, N.S.; Flake, A.W.; Hedrick, H.L. Fetal intrapericardial teratoma: Natural history and management including successful in utero surgery. Am. J. Obstet. Gynecol. 2016, 215, 780.e1–780.e7.
- Srisupundit, K.; Sukpan, K.; Tongsong, T.; Traisrisilp, K. Prenatal sonographic features of fetal mediastinal teratoma. J. Clin. Ultrasound 2020, 48, 419–422.
- Wang, B.; Feng, Y.; Guo, Y.; Kan, Q.; Zou, Y.; Wu, Y.; Zheng, M.; Cheng, R. Clinical features and outcomes of congenital chylothorax: A single tertiary medical center experience in China. J. Cardiothorac. Surg. 2022, 17, 276.
- Sekar, P.; Hornberger, L.K. The role of fetal echocardiography in fetal intervention: A symbiotic relationship. Clin. Perinatol. 2009, 36, 301–327.
More