A wide range of histological as well as clinical properties are exhibited by B-cell non-Hodgkin’s lymphomas. These properties could make the diagnostics process complicated. The diagnosis of lymphomas at an initial stage is essential because early remedial actions taken against destructive subtypes are commonly deliberated as successful and restorative. New possibilities are now open for diagnosing cancer with the help of metabolomics. The study of all the metabolites synthesised in the human body is called “metabolomics.” A patient’s phenotype is directly linked with metabolomics, which can help in providing some clinically beneficial biomarkers and is applied in the diagnostics of B-cell non-Hodgkin’s lymphoma. In cancer research, it can analyse the cancerous metabolome to identify the metabolic biomarkers.
- metabolomics
- B-cell non-Hodgkin’s lymphoma
- biomarkers
- metabolites
- early diagnosis
1. Introduction
2. Metabolism in B-Cell Non-Hodgkin’s Lymphoma (B-NHL)
Cell metabolism is a well-defined set of metabolic activities that generate and store energy equivalents, maintain redox homeostasis, synthesise biologically active macromolecules, and eliminate organic waste [17][15]. Catabolism breaks down carbon sources into simpler intermediates, which are then employed as building blocks in the production of lipids, amino acids, carbohydrates, and nucleotides (anabolism) [18][16]. Tumour cells are able to survive, grow, and divide because of their metabolic versatility and plasticity, which allow them to produce ATP as an energy source while maintaining the reduction–oxidation (redox) balance and devoting resources to biosynthesis [19][17]. Metabolic alterations in B-NHL are characterised by the production of enough energy and maintenance of anabolism for survival, growth, and division in the face of low levels of nutrients and oxygen (such as HIF1 and MYC), deregulation of metabolic regulators (like mTORC1), and rewiring of metabolic pathways (e.g., BCR signalling) [22,23][18][19]. The Warburg effect promotes aerobic glycolysis over aerobic oxidation [24][20], and this is supported by HIF1-alpha and MYC. This leads to the production of lactate and poor producing ATP, but helps create biomass. As a result, the body’s reaction to hypoxia-induced metabolic abnormalities may promote anabolism in GC-derived B-cell lymphoma [22][18]. MYC oncogene aberrations, including translocations or overexpression, are characteristics of B-cell lymphoma aetiology [25][21]. B-cell lymphomas require higher MYC levels to maintain their rapid proliferation rate. MYC upregulates nucleoside metabolism, which is essential for cell development. Glutamine metabolism is similarly regulated by MYC expression [25,26][21][22]. Glucose uptake, glycolysis, and lipid biosynthesis are all controlled by MYC as well [27][23]. On the other hand, alpha-ketoglutarate (αKG) synthesis can be inhibited by hypoxia and mitochondrial dysfunction, which in turn reduces the activity of αKG-dependent enzymes, leading to increased DNA and histone hypermethylation and stabilisation of HIF1α. HIF1α is the primary transcriptional regulator of the adaptive response to hypoxia and is constitutively stabilised in a significant proportion of DLBCLs and FLs [22][18]. HIF1α and MYC promote anaerobic glycolysis by activating genes for glucose transporter (GLUT), hexokinase (HK), monocarboxylate transporter (MTC), pyruvate dehydrogenase (PDK), phosphofructokinase (PFK), phosphoglycerate kinase (PGK), pyruvate kinase (PK), and lactate dehydrogenase (LDHA) [27][23]. mTORC1 is essential for generating metabolic precursors via the tricarboxylic acid cycle (TCA) and stimulating cellular proliferation. Activation of mTORC1 thereby enhances the survival of B-cell lymphoma. T-cell-selected GC B cells in the light zone necessitate mTORC1 activation in order to proliferate and mutate in the dark zone. mTORC1 may be aberrantly activated in GCB-DLBCL through activating mutations of PI3K/Akt/mTOR pathway genes [22][18]. A further marker of B-cell lymphoma is altered B-cell receptor (BCR) signalling, which is essential for the maintenance and creation of both healthy and malignant B cells [28][24]. PI3K/AKT/mTORC1 is one of the BCR signalling pathway’s downstream branches. PI3K regulates glycolysis and energy generation, and consequent AKT signalling influences the cellular metabolome. AKT promotes glucose uptake and glycolysis by increasing the expression and translocation of GLUT1 and glycolytic enzymes, including hexokinase (HK) expression and activation [28][24]. In a subset of DLBCL and MCL, PTEN mutations lead to AKT/mTORC1 pathway gene expression [29][25]. RagC mutations in FL enhance mTORC1 signalling by eliminating amino acid dependence [30][26]. Numerous anabolic and energy-generating processes, including protein synthesis, pyrimidine synthesis, HIF1α expression, glycolysis, the oxidative portion of the pentose phosphate pathway (PPP), lipid and mitochondrial metabolism, and glutaminolysis, are stimulated by mTORC1 expression [23][19]. There is an urgent need for biomarkers based on non-invasive sampling procedures (e.g., blood, urine, etc.) that can help in the diagnosis of lymphoma, such as metabolite profiling. The perfect test should be easy, reliable, and accurate. “What simple, non-invasive, painless, and convenient tests can be used to detect cancer early?” ranked as the most important research priority for the early detection of cancer in the UK-focused research gap survey performed by the James Lind Alliance, which includes patients and doctors [31][27] (Figure 1).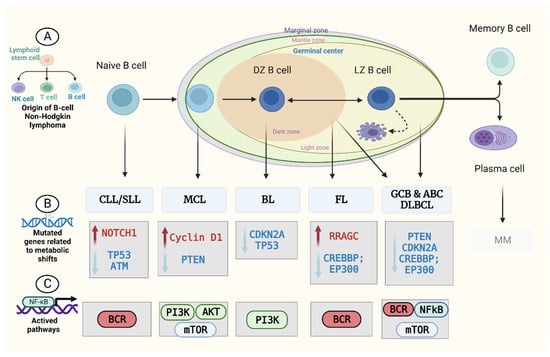
3. Metabolomics and B-NHL Biomarker Discovery
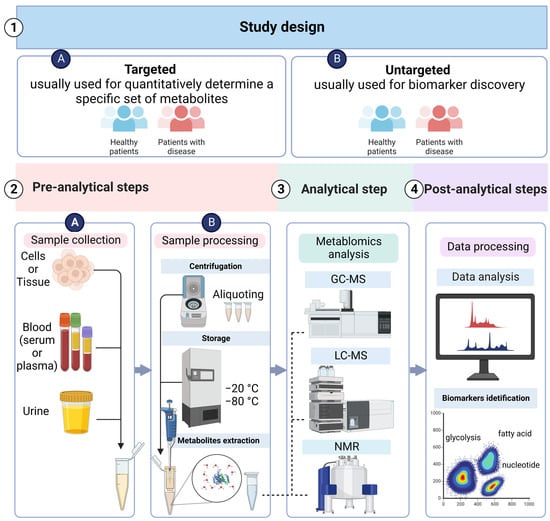
3.1. Metabolomics Study Design
3.2. Sample Collection and Preparation
The collection of the sample, its preparation, and storage are the second step in the metabolomics study plan. The most common samples for conducting clinical metabolomics research are blood and urine [88][39]. It is important to design the research based on metabolomics to reduce the influence of certain constituents such as age, gender, state of fasting, diet, physical activity, exercise, and the day and time of sample collection. Before starting the actual research, it is important to conduct a pilot study of healthy individuals and report it as part of the research to validate the results’ reproducibility. The samples (particularly plasma, serum, and urine) must be kept in various aliquots soon after collection to avoid the production of compounds from the many freeze–thaw cycles used for different metabolomics studies [89][40]. The factors used for processing the sample, such as pH buffering and extraction, should also be uniform and follow standard operating procedures (SOPs) [81,82,90,91][35][36][41][42]. The samples that are non-invasive in nature, such as blood or urine, are the best for regular clinical analysis [85][43].3.3. Analytical Techniques
3.3.1. LC–MS
The MS technique has the ability to isolate the intricate mixture of compounds for their detection and quantification with elevated sensitivity and specificity, and can also demonstrate information regarding molecular structures [99][44]. MS separation techniques are essential for reducing sample complexity and minimising ionisation suppression effects [100][45]. A preceding separation stage, such as high-performance liquid chromatography (HPLC), or ultra-performance liquid chromatography (UPLC), and capillary electrophoresis (CE), is frequently required.3.3.2. GC–MS
GC–MS is a technique that combines great separation efficacy with sensitive, selective, and versatile mass evaluation and is suitable for comprehensive analysis. It is a combination of MS and GS that is used for the detection and quantification of a wide range of chemical compounds, such as natural products, blood, and urine. GS–MS is used in many fields of study, such as detecting drugs, amino acid evaluation, doping control, and the detection of natural materials like food products [103][46]. EI, or electron ionization, is used for combining MS with GC in almost all the metabolomics applications that are based on GC. The EI–MS method works well for chemical compounds that do not change when heated and that are volatile and are separated by chromatography at high temperatures [104][47].3.3.3. NMR
NMR spectroscopy is a universal metabolite detection method that allows for direct analysis of samples with little sample preparation and simultaneous measurement of numerous types of tiny metabolites [106,107][48][49]. However, it has limitations, such as high equipment costs, high maintenance costs, and decreased sensitivity [108,109][50][51]. Mass spectrometry is better than NMR in several ways, although NMR has its own advantages.3.4. Data Acquisition and Processing
When the metabolomics data are produced, it is important to ensure that they are reproducible [89,110][40][52]. Quality standardisation and quality control are considered for the optimisation of the reproducibility of results. Data analysis and bioinformatics are used to process the data, which are then subjected to statistical analysis. There are two classical approaches to the statistical analysis of multivariate data: unsupervised learning and supervised learning. A popular unsupervised learning method is principal component analysis (PCA). The second main approach is supervised learning, such as with artificial neural networks (ANN), partial least squares discriminate analysis (PLS-DA), etc., which can be used for excavating the data further to obtain the biomarkers [111,112][53][54]. The discovery process of biomarkers can be driven through supervised models that can be linked with clinical results, histopathological scores, and various other omics data. It is important to test the supervised models with precise internal cross-validation processes or external tests to obtain trusted biomarkers and models and to decrease the chances of data overfitting [113][55].3.5. Metabolites Identification: Biomarker Discovery and Validation
Profiling the metabolites in each biological entity is incomplete without accurate data measurement and precise interpretation. To identify the features of potent spectral biomarkers, attempts are made to recognise the unidentified spectral biomarkers. The peaks can be identified with the help of public metabolomics databases and in-house spectral databases such as the Golm database, LIPID MAPS, human metabolome database (HMDB), METLIN database, etc. Following the identification of metabolomics biomarkers, additional experiments are required to validate or test the biomarkers [82,112,114][36][54][56].4. Applications of Metabolomics in B-NHL
4.1. Discovering Targeted Therapies Based on Metabolomics
Metabolism in B-NHL plays a crucial role in established therapeutic approaches. Antimetabolites were the name given to the chemical compounds that were first used to treat cancer. The reason for choosing this name was that these compounds were found to resemble endogenous metabolites in their chemical structure and disrupt the process of normal metabolism. In comparison to other omics, metabolomics is best for evaluating the potential of these cancer treatment regimens. The study was carried out to discover whether the therapies could cause alterations in the metabolic pathways and detect the pharmacokinetics of drugs simultaneously or not. In the coming time, it will become crucial to combine the study of pharmacometabolomics with other biological systems knowledge, such as mRNA, genetics, miRNA, and imaging. This will help in determining the correlation of the metabolomics response with the cancer stage, undesirable incidents, and the growth or recession of the tumour. The study of pharmacometabolomics is capable of monitoring a patient’s metabolic response to a drug; thus, it is very interesting to use metabolomics in detecting cancerous growth, prognosis, and therapy management [82][36].4.2. Determining B-NHL Diagnostic and Prognostic Biomarkers
A recent metabolomics study suggested a methodology for discovering novel biomarkers that can be used for the diagnosis and characterisation of various lymphoma subtypes. The GC–MS method was used for the investigation and evaluation of plasma samples taken from individuals with different subtypes of lymphomas. The results showed a significant prevalence of elaidic acid and hypoxanthine (HX) in patients suffering from Hodgkin’s lymphoma, MM, CLL, and DLBCL compared with healthy control individuals in all the study groups [50][57].4.3. Determining the Lymphomagenesis Risk Factors
Genetic mutations accumulate sequentially during tumour development, eventually resulting in malignant tumours. However, it has also been shown that metabolic processes and inflammatory factors indirectly contribute to the development of the tumour [11][58]. In their study, Pettersena et al. proved that the cell line of B-cell lymphoma surrounds numerous amplified genomic uracil concentrations in comparison with non-lymphoma cell lines or normal lymphocytes. They utilised a method based on liquid chromatography combined with mass spectrometry (LC/MS) for quantifying the genomic sequence of 2-deoxyuridine and proving their study. In harmony with uracil generated by activation-induced cytidine deaminase (AID), they discovered a distinctive mutational signature of an AID hotspot in the lymphoma area where there was clustered mutation. They also presented an important revelation about the expression of SMUG1 and uracil-DNA glycosylases UNG along with the excision capacity of uracil by stating its negative correlation with the concentration of genomic uracil, which somewhat decreased the AID effect [129][59]. Another study was also conducted on the metabolomic pattern of Burkitt lymphoma that was induced by MYC glucose deprivation, as well as hypoxic and aerobic conditions. They used a [U-13C, 15N]-glutamine tracer to detect glutamine import and metabolism via the TCA cycle under hypoxia conditions and discovered that glutamine is significantly precipitated to citrate carbons. The deficiency of glucose leads to the significant augmentation of citrate, fumarate, and glutamine-derived malate. Their arrangements showed a different pathway for the generation of energy called glutaminolysis, which is associated with the glucose-independent TCA cycle. Under the conditions of hypoxia and scarcity of glucose, the critical role of glutamine in the proliferation of cells makes them susceptible to BPTES (glutaminase inhibitors), which in turn can be used for treating tumours [130][60].References
- Szymańska, K.; Park, S. Non-Hodgkin Lymphoma: Diagnosis and Treatment. In Reference Module in Biomedical Sciences; Elsevier: Amstedam, The Netherlands, 2018. [Google Scholar] [CrossRef]Szymańska, K.; Park, S. Non-Hodgkin Lymphoma: Diagnosis and Treatment. In Reference Module in Biomedical Sciences; Elsevier: Amstedam, The Netherlands, 2018.
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood J. Am. Soc. Hematol. 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed][Green Version]Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood J. Am. Soc. Hematol. 2016, 127, 2375–2390.
- Pophali, P.; Marinelli, L.M.; Ketterling, R.P.; Meyer, R.G.; McPhail, E.D.; Kurtin, P.J.; Habermann, T.M.; King, R.L. High Level MYC Amplification in Aggressive B-Cell Lymphomas: Is It a Marker of Aggressive Disease? Blood 2018, 132 (Suppl. 1), 1693. [Google Scholar] [CrossRef]Pophali, P.; Marinelli, L.M.; Ketterling, R.P.; Meyer, R.G.; McPhail, E.D.; Kurtin, P.J.; Habermann, T.M.; King, R.L. High Level MYC Amplification in Aggressive B-Cell Lymphomas: Is It a Marker of Aggressive Disease? Blood 2018, 132 (Suppl. 1), 1693.
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef] [PubMed]Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446.
- Cai, W.; Zeng, Q.; Zhang, X.; Ruan, W. Trends Analysis of Non-Hodgkin Lymphoma at the National, Regional, and Global Level 1990–2019: Results from the Global Burden of Disease Study 2019. Front. Med. 2021, 8. [Google Scholar] [CrossRef] [PubMed]Cai, W.; Zeng, Q.; Zhang, X.; Ruan, W. Trends Analysis of Non-Hodgkin Lymphoma at the National, Regional, and Global Level 1990–2019: Results from the Global Burden of Disease Study 2019. Front. Med. 2021, 8.
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic Regulation of T Lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef][Green Version]MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic Regulation of T Lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283.
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef][Green Version]Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47.
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef][Green Version]Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669.
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef][Green Version]Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180.
- Newman, J.S.; Francis, I.R.; Kaminski, M.S.; Wahl, R.L. Imaging of Lymphoma with PET with 2-[F-18]-Fluoro-2-Deoxy-D-Glucose: Correlation with CT. Radiology 1994, 190, 111–116. [Google Scholar] [CrossRef]Zhou, J.; Yu, S.; Wang, Y.; Gu, X.; Wu, Q.; Xue, Y.; Shan, G.; Zhang, H.; Zhao, W.; Yan, C. Serum Metabolite Profiling of B-Cell Non-Hodgkin’s Lymphoma Using UPLC-QTOFMS and GC-TOFMS. Metabolomics 2014, 10, 677–687.
- Schmidt, D.R.; Patel, R.; Kirsch, D.G.; Lewis, C.A.; Vander Heiden, M.G.; Locasale, J.W. Metabolomics in Cancer Research and Emerging Applications in Clinical Oncology. CA. Cancer J. Clin. 2021, 71, 333–358. [Google Scholar] [CrossRef]Wang, Y.; Zhang, L.; Chen, W.L.; Wang, J.H.; Li, N.; Li, J.M.; Mi, J.Q.; Zhang, W.N.; Li, Y.; Wu, S.F.; et al. Rapid Diagnosis and Prognosis of de Novo Acute Myeloid Leukemia by Serum Metabonomic Analysis. J. Proteome Res. 2013, 12, 4393–4401.
- Zhou, J.; Yu, S.; Wang, Y.; Gu, X.; Wu, Q.; Xue, Y.; Shan, G.; Zhang, H.; Zhao, W.; Yan, C. Serum Metabolite Profiling of B-Cell Non-Hodgkin’s Lymphoma Using UPLC-QTOFMS and GC-TOFMS. Metabolomics 2014, 10, 677–687. [Google Scholar] [CrossRef]Denkert, C.; Bucher, E.; Hilvo, M.; Salek, R.; Orešič, M.; Griffin, J.; Brockmöller, S.; Klauschen, F.; Loibl, S.; Barupal, D.K.; et al. Metabolomics of Human Breast Cancer: New Approaches for Tumor Typing and Biomarker Discovery. Genome Med. 2012, 4, 37.
- Wang, Y.; Zhang, L.; Chen, W.L.; Wang, J.H.; Li, N.; Li, J.M.; Mi, J.Q.; Zhang, W.N.; Li, Y.; Wu, S.F.; et al. Rapid Diagnosis and Prognosis of de Novo Acute Myeloid Leukemia by Serum Metabonomic Analysis. J. Proteome Res. 2013, 12, 4393–4401. [Google Scholar] [CrossRef] [PubMed]Hilvo, M.; Denkert, C.; Lehtinen, L.; Müller, B.; Brockmöller, S.; Seppänen-Laakso, T.; Budczies, J.; Bucher, E.; Yetukuri, L.; Castillo, S.; et al. Novel Theranostic Opportunities Offered by Characterization of Altered Membrane Lipid Metabolism in Breast Cancer Progression. Cancer Res. 2011, 71, 3236–3245.
- Denkert, C.; Bucher, E.; Hilvo, M.; Salek, R.; Orešič, M.; Griffin, J.; Brockmöller, S.; Klauschen, F.; Loibl, S.; Barupal, D.K.; et al. Metabolomics of Human Breast Cancer: New Approaches for Tumor Typing and Biomarker Discovery. Genome Med. 2012, 4, 37. [Google Scholar] [CrossRef] [PubMed][Green Version]Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of Lactate Dehydrogenase a Induces Oxidative Stress and Inhibits Tumor Progression. PNAS 2010, 107, 2037–2042.
- Hilvo, M.; Denkert, C.; Lehtinen, L.; Müller, B.; Brockmöller, S.; Seppänen-Laakso, T.; Budczies, J.; Bucher, E.; Yetukuri, L.; Castillo, S.; et al. Novel Theranostic Opportunities Offered by Characterization of Altered Membrane Lipid Metabolism in Breast Cancer Progression. Cancer Res. 2011, 71, 3236–3245. [Google Scholar] [CrossRef][Green Version]Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033.
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of Lactate Dehydrogenase a Induces Oxidative Stress and Inhibits Tumor Progression. PNAS 2010, 107, 2037–2042. [Google Scholar] [CrossRef][Green Version]DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200.
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef][Green Version]Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer Metabolism: A Therapeutic Perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31.
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef][Green Version]Mlynarczyk, C.; Fontán, L.; Melnick, A. Germinal Center-derived Lymphomas: The Darkest Side of Humoral Immunity. Immunol. Rev. 2019, 288, 214–239.
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer Metabolism: A Therapeutic Perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]Beielstein, A.C.; Pallasch, C.P. Tumor Metabolism as a Regulator of Tumor–Host Interactions in the B-Cell Lymphoma Microenvironment—Fueling Progression and Novel Brakes for Therapy. Int. J. Mol. Sci. 2019, 20, 4158.
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular Subtypes of Diffuse Large B Cell Lymphoma Are Associated with Distinct Pathogenic Mechanisms and Outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef]Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314.
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations Driving CLL and Their Evolution in Progression and Relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef][Green Version]Dejure, F.R.; Eilers, M. MYC and Tumor Metabolism: Chicken and Egg. EMBO J. 2017, 36, 3409–3420.
- Mlynarczyk, C.; Fontán, L.; Melnick, A. Germinal Center-derived Lymphomas: The Darkest Side of Humoral Immunity. Immunol. Rev. 2019, 288, 214–239. [Google Scholar] [CrossRef][Green Version]Dejure, F.R.; Royla, N.; Herold, S.; Kalb, J.; Walz, S.; Ade, C.P.; Mastrobuoni, G.; Vanselow, J.T.; Schlosser, A.; Wolf, E.; et al. The MYC MRNA 3′-UTR Couples RNA Polymerase II Function to Glutamine and Ribonucleotide Levels. EMBO J. 2017, 36, 1854–1868.
- Beielstein, A.C.; Pallasch, C.P. Tumor Metabolism as a Regulator of Tumor–Host Interactions in the B-Cell Lymphoma Microenvironment—Fueling Progression and Novel Brakes for Therapy. Int. J. Mol. Sci. 2019, 20, 4158. [Google Scholar] [CrossRef] [PubMed][Green Version]Broecker-Preuss, M.; Becher-Boveleth, N.; Bockisch, A.; Dührsen, U.; Müller, S. Regulation of Glucose Uptake in Lymphoma Cell Lines by C-MYC- and PI3K-Dependent Signaling Pathways and Impact of Glycolytic Pathways on Cell Viability. J. Transl. Med. 2017, 15, 158.
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]Vangapandu, H.V.; Havranek, O.; Ayres, M.L.; Kaipparettu, B.A.; Balakrishnan, K.; Wierda, W.G.; Keating, M.J.; Davis, R.E.; Stellrecht, C.M.; Gandhi, V. B-Cell Receptor Signaling Regulates Metabolism in Chronic Lymphocytic Leukemia. Mol. Cancer Res. 2017, 15, 1692–1703.
- Dejure, F.R.; Eilers, M. MYC and Tumor Metabolism: Chicken and Egg. EMBO J. 2017, 36, 3409–3420. [Google Scholar] [CrossRef]Wang, X.; Cao, X.; Sun, R.; Tang, C.; Tzankov, A.; Zhang, J.; Manyam, G.C.; Xiao, M.; Miao, Y.; Jabbar, K.; et al. Clinical Significance of PTEN Deletion, Mutation, and Loss of PTEN Expression in De Novo Diffuse Large B-Cell Lymphoma. Neoplasia 2018, 20, 574–593.
- Dejure, F.R.; Royla, N.; Herold, S.; Kalb, J.; Walz, S.; Ade, C.P.; Mastrobuoni, G.; Vanselow, J.T.; Schlosser, A.; Wolf, E.; et al. The MYC MRNA 3′-UTR Couples RNA Polymerase II Function to Glutamine and Ribonucleotide Levels. EMBO J. 2017, 36, 1854–1868. [Google Scholar] [CrossRef] [PubMed]Okosun, J.; Wolfson, R.L.; Wang, J.; Araf, S.; Wilkins, L.; Castellano, B.M.; Escudero-Ibarz, L.; Al Seraihi, A.F.; Richter, J.; Bernhart, S.H.; et al. Recurrent MTORC1-Activating RRAGC Mutations in Follicular Lymphoma. Nat. Genet. 2016, 48, 183–188.
- Broecker-Preuss, M.; Becher-Boveleth, N.; Bockisch, A.; Dührsen, U.; Müller, S. Regulation of Glucose Uptake in Lymphoma Cell Lines by C-MYC- and PI3K-Dependent Signaling Pathways and Impact of Glycolytic Pathways on Cell Viability. J. Transl. Med. 2017, 15, 158. [Google Scholar] [CrossRef] [PubMed][Green Version]Badrick, E.; Cresswell, K.; Ellis, P.; Renehan, A.G.; Crosbie, E.J.; Crosbie, P.; Hall, P.S.; O’Flynn, H.; Martin, R.; Leighton, J.; et al. Top Ten Research Priorities for Detecting Cancer Early. Lancet Public Health 2019, 4, e551.
- Vangapandu, H.V.; Havranek, O.; Ayres, M.L.; Kaipparettu, B.A.; Balakrishnan, K.; Wierda, W.G.; Keating, M.J.; Davis, R.E.; Stellrecht, C.M.; Gandhi, V. B-Cell Receptor Signaling Regulates Metabolism in Chronic Lymphocytic Leukemia. Mol. Cancer Res. 2017, 15, 1692–1703. [Google Scholar] [CrossRef] [PubMed][Green Version]Shaffer, A.L.; Young, R.M.; Staudt, L.M. Pathogenesis of Human B Cell Lymphomas. Annu. Rev. Immunol. 2012, 30, 565–610.
- Wang, X.; Cao, X.; Sun, R.; Tang, C.; Tzankov, A.; Zhang, J.; Manyam, G.C.; Xiao, M.; Miao, Y.; Jabbar, K.; et al. Clinical Significance of PTEN Deletion, Mutation, and Loss of PTEN Expression in De Novo Diffuse Large B-Cell Lymphoma. Neoplasia 2018, 20, 574–593. [Google Scholar] [CrossRef] [PubMed]Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M.; et al. Whole-Genome Sequencing Identifies Recurrent Mutations in Chronic Lymphocytic Leukaemia. Nature 2011, 475, 101–105.
- Okosun, J.; Wolfson, R.L.; Wang, J.; Araf, S.; Wilkins, L.; Castellano, B.M.; Escudero-Ibarz, L.; Al Seraihi, A.F.; Richter, J.; Bernhart, S.H.; et al. Recurrent MTORC1-Activating RRAGC Mutations in Follicular Lymphoma. Nat. Genet. 2016, 48, 183–188. [Google Scholar] [CrossRef][Green Version]Zenz, T.; Mertens, D.; Küppers, R.; Döhner, H.; Stilgenbauer, S. From Pathogenesis to Treatment of Chronic Lymphocytic Leukaemia. Nat. Rev. Cancer 2010, 10, 37–50.
- Badrick, E.; Cresswell, K.; Ellis, P.; Renehan, A.G.; Crosbie, E.J.; Crosbie, P.; Hall, P.S.; O’Flynn, H.; Martin, R.; Leighton, J.; et al. Top Ten Research Priorities for Detecting Cancer Early. Lancet Public Health 2019, 4, e551. [Google Scholar] [CrossRef][Green Version]Pérez-Galán, P.; Mora-Jensen, H.; Weniger, M.A.; Shaffer, A.L.; Rizzatti, E.G.; Chapman, C.M.; Mo, C.C.; Stennett, L.S.; Rader, C.; Liu, P.; et al. Bortezomib Resistance in Mantle Cell Lymphoma Is Associated with Plasmacytic Differentiation. Blood 2011, 117, 542–552.
- Bryan, J.N. The Current State of Clinical Application of Serum Biomarkers for Canine Lymphoma. Front. Vet. Sci. 2016, 3. [Google Scholar] [CrossRef] [PubMed][Green Version]Ramis-Zaldivar, J.E.; Gonzalez-Farré, B.; Balagué, O.; Celis, V.; Nadeu, F.; Salmerón-Villalobos, J.; Andrés, M.; Martin-Guerrero, I.; Garrido-Pontnou, M.; Gaafar, A.; et al. Distinct Molecular Profile of IRF4-Rearranged Large B-Cell Lymphoma. Blood 2020, 135, 274–286.
- Shaffer, A.L.; Young, R.M.; Staudt, L.M. Pathogenesis of Human B Cell Lymphomas. Annu. Rev. Immunol. 2012, 30, 565–610. [Google Scholar] [CrossRef] [PubMed]Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating Mutations of Acetyltransferase Genes in B-Cell Lymphoma. Nature 2011, 471, 189–195.
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M.; et al. Whole-Genome Sequencing Identifies Recurrent Mutations in Chronic Lymphocytic Leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef] [PubMed][Green Version]Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the Coding Genome of Diffuse Large B-Cell Lymphoma. Nat. Genet. 2011, 43, 830–837.
- Zenz, T.; Mertens, D.; Küppers, R.; Döhner, H.; Stilgenbauer, S. From Pathogenesis to Treatment of Chronic Lymphocytic Leukaemia. Nat. Rev. Cancer 2010, 10, 37–50. [Google Scholar] [CrossRef]Mamas, M.; Dunn, W.B.; Neyses, L.; Goodacre, R. The Role of Metabolites and Metabolomics in Clinically Applicable Biomarkers of Disease. Arch. Toxicol. 2011, 85, 5–17.
- Pérez-Galán, P.; Mora-Jensen, H.; Weniger, M.A.; Shaffer, A.L.; Rizzatti, E.G.; Chapman, C.M.; Mo, C.C.; Stennett, L.S.; Rader, C.; Liu, P.; et al. Bortezomib Resistance in Mantle Cell Lymphoma Is Associated with Plasmacytic Differentiation. Blood 2011, 117, 542–552. [Google Scholar] [CrossRef][Green Version]Beger, R.D. A Review of Applications of Metabolomics in Cancer. Metabolites 2013, 3, 552.
- Ramis-Zaldivar, J.E.; Gonzalez-Farré, B.; Balagué, O.; Celis, V.; Nadeu, F.; Salmerón-Villalobos, J.; Andrés, M.; Martin-Guerrero, I.; Garrido-Pontnou, M.; Gaafar, A.; et al. Distinct Molecular Profile of IRF4-Rearranged Large B-Cell Lymphoma. Blood 2020, 135, 274–286. [Google Scholar] [CrossRef]Nalbantoglu, S. Metabolomics: Basic Principles and Strategies. In Molecular Medicine; IntechOpen: London, UK, 2019.
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating Mutations of Acetyltransferase Genes in B-Cell Lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef][Green Version]Carneiro, G.; Radcenco, A.L.; Evaristo, J.; Monnerat, G. Novel Strategies for Clinical Investigation and Biomarker Discovery: A Guide to Applied Metabolomics. Horm. Mol. Biol. Clin. Investig. 2019, 38.
- Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the Coding Genome of Diffuse Large B-Cell Lymphoma. Nat. Genet. 2011, 43, 830–837. [Google Scholar] [CrossRef][Green Version]Spratlin, J.L.; Serkova, N.J.; Eckhardt, S.G. Clinical Applications of Metabolomics in Oncology: A Review. Clin. Cancer Res. 2009, 15, 431–440.
- Xie, Y.; Pittaluga, S.; Jaffe, E.S. The Histological Classification of Diffuse Large B-Cell Lymphomas. Semin. Hematol. 2015, 52, 57–66. [Google Scholar] [CrossRef][Green Version]Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for Large-Scale Metabolic Profiling of Serum and Plasma Using Gas Chromatography and Liquid Chromatography Coupled to Mass Spectrometry. Nat. Protoc. 2011, 6, 1060–1083.
- Flowers, C.R.; Sinha, R.; Vose, J.M. Improving Outcomes for Patients with Diffuse Large B-Cell Lymphoma. CA. Cancer J. Clin. 2010, 60, 393–408. [Google Scholar] [CrossRef]Ganti, S.; Weiss, R.H. Urine Metabolomics for Kidney Cancer Detection and Biomarker Discovery. Urol. Oncol. Semin. Orig. Investig. 2011, 29, 551–557.
- Ninan, M.J.; Wadhwa, P.D.; Gupta, P. Prognostication of Diffuse Large B-Cell Lymphoma in the Rituximab Era. Leuk. Lymphoma 2011, 52, 360–373. [Google Scholar] [CrossRef] [PubMed]Nielsen, T.H.; Diaz, Z.; Christodoulopoulos, R.; Charbonneau, F.; Qureshi, S.; Rousseau, C.; Benlimame, N.; Camlioglu, E.; Constantin, A.M.; Oros, K.K.; et al. Methods for Sample Acquisition and Processing of Serial Blood and Tumor Biopsies for Multicenter Diffuse Large B-Cell Lymphoma Clinical Trials. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 2688–2693.
- Morita, N.; Hoshi, M.; Hara, T.; Ninomiya, S.; Enoki, T.; Yoneda, M.; Tsurumi, H.; Saito, K. Viability of Diffuse Large B-Cell Lymphoma Cells Is Regulated by Kynurenine 3-Monooxygenase Activity. Oncol. Lett. 2021, 22, 790. [Google Scholar] [CrossRef] [PubMed]Jacob, M.; Lopata, A.L.; Dasouki, M.; Abdel Rahman, A.M. Metabolomics toward Personalized Medicine. Mass Spectrom. Rev. 2019, 38, 221–238.
- Bhalla, K.; Jaber, S.; Nahid, M.N.; Underwood, K.; Beheshti, A.; Landon, A.; Bhandary, B.; Bastian, P.; Evens, A.M.; Haley, J.; et al. Role of Hypoxia in Diffuse Large B-Cell Lymphoma: Metabolic Repression and Selective Translation of HK2 Facilitates Development of DLBCL. Sci. Rep. 2018, 8, 744. [Google Scholar] [CrossRef] [PubMed][Green Version]Pitt, J.J. Principles and Applications of Liquid Chromatography-Mass Spectrometry in Clinical Biochemistry. Clin. Biochem. Rev. 2009, 30, 19–34.
- Chiche, J.; Reverso-Meinietti, J.; Mouchotte, A.; Rubio-Patiño, C.; Mhaidly, R.; Villa, E.; Bossowski, J.P.; Proics, E.; Grima-Reyes, M.; Paquet, A.; et al. GAPDH Expression Predicts the Response to R-CHOP, the Tumor Metabolic Status, and the Response of DLBCL Patients to Metabolic Inhibitors. Cell Metab. 2019, 29, 1243–1257.e10. [Google Scholar] [CrossRef] [PubMed]Kuehnbaum, N.L.; Britz-McKibbin, P. New Advances in Separation Science for Metabolomics: Resolving Chemical Diversity in a Post-Genomic Era. Chem. Rev. 2013, 113, 2437–2468.
- Eraslan, Z.; Papatzikas, G.; Cazier, J.-B.; Khanim, F.L.; Günther, U.L. Targeting Asparagine and Serine Metabolism in Germinal Centre-Derived B Cells Non-Hodgkin Lymphomas (B-NHL). Cells 2021, 10, 2589. [Google Scholar] [CrossRef]Fiehn, O. Metabolomics by Gas Chromatography–Mass Spectrometry: Combined Targeted and Untargeted Profiling. Curr. Protoc. Mol. Biol. 2016, 114.
- Choueiry, F.; Singh, S.; Sircar, A.; Laliotis, G.; Sun, X.; Chavdoula, E.; Zhang, S.; Helmig-Mason, J.; Hart, A.; Epperla, N.; et al. Integration of Metabolomics and Gene Expression Profiling Elucidates IL4I1 as Modulator of Ibrutinib Resistance in ABC-Diffuse Large B Cell Lymphoma. Cancers 2021, 13, 2146. [Google Scholar] [CrossRef]Koek, M.M.; Jellema, R.H.; van der Greef, J.; Tas, A.C.; Hankemeier, T. Quantitative Metabolomics Based on Gas Chromatography Mass Spectrometry: Status and Perspectives. Metabolomics 2011, 7, 307–328.
- Stenson, M.; Pedersen, A.; Hasselblom, S.; Nilsson-Ehle, H.; Karlsson, B.G.; Pinto, R.; Andersson, P.-O. Serum Nuclear Magnetic Resonance-Based Metabolomics and Outcome in Diffuse Large B-Cell Lymphoma Patients—A Pilot Study. Leuk. Lymphoma 2016, 57, 1814–1822. [Google Scholar] [CrossRef]Lane, A.N.; Fan, T.W.-M. NMR-Based Stable Isotope Resolved Metabolomics in Systems Biochemistry. Arch. Biochem. Biophys. 2017, 628, 123–131.
- Mi, M.; Liu, Z.; Zheng, X.; Wen, Q.; Zhu, F.; Li, J.; Mungur, I.D.; Zhang, L. Serum Metabolomic Profiling Based on GC/MS Helped to Discriminate Diffuse Large B-Cell Lymphoma Patients with Different Prognosis. Leuk. Res. 2021, 111, 106693. [Google Scholar] [CrossRef]Fan, T.W.-M.; Lane, A.N. Applications of NMR Spectroscopy to Systems Biochemistry. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 92–93, 18–53.
- Barberini, L.; Noto, A.; Fattuoni, C.; Satta, G.; Zucca, M.; Cabras, M.G.; Mura, E.; Cocco, P. The Metabolomic Profile of Lymphoma Subtypes: A Pilot Study. Molecules 2019, 24, 13. [Google Scholar] [CrossRef][Green Version]Nagana Gowda, G.A.; Raftery, D. Can NMR Solve Some Significant Challenges in Metabolomics? J. Magn. Reson. 2015, 260, 144–160.
- Zheng, M.; Zhou, X.; Wang, Q.; Chen, X.; Cao, B.; Li, J. Metabolomic Approach to Characterize the Metabolic Phenotypes and Varied Response to Ouabain of Diffuse Large B-Cell Lymphoma Cells. Leuk. Lymphoma 2021, 62, 1597–1608. [Google Scholar] [CrossRef]Tiziani, S.; Kang, Y.; Choi, J.S.; Roberts, W.; Paternostro, G. Metabolomic High-Content Nuclear Magnetic Resonance-Based Drug Screening of a Kinase Inhibitor Library. Nat. Commun. 2011, 2, 545.
- Schwarzfischer, P.; Reinders, J.; Dettmer, K.; Kleo, K.; Dimitrova, L.; Hummel, M.; Feist, M.; Kube, D.; Szczepanowski, M.; Klapper, W.; et al. Comprehensive Metaboproteomics of Burkitt’s and Diffuse Large B-Cell Lymphoma Cell Lines and Primary Tumor Tissues Reveals Distinct Differences in Pyruvate Content and Metabolism. J. Proteome Res. 2017, 16, 1105–1120. [Google Scholar] [CrossRef] [PubMed]Dunn, W.B.; Wilson, I.D.; Nicholls, A.W.; Broadhurst, D. The Importance of Experimental Design and QC Samples in Large-Scale and MS-Driven Untargeted Metabolomic Studies of Humans. Bioanalysis 2012, 4, 2249–2264.
- Fei, F.; Zheng, M.; Xu, Z.; Sun, R.; Chen, X.; Cao, B.; Li, J. Plasma Metabolites Forecast Occurrence and Prognosis for Patients with Diffuse Large B-Cell Lymphoma. Front. Oncol. 2022, 12. [Google Scholar] [CrossRef] [PubMed]Fonville, J.M.; Richards, S.E.; Barton, R.H.; Boulange, C.L.; Ebbels, T.M.D.; Nicholson, J.K.; Holmes, E.; Dumas, M.-E. The Evolution of Partial Least Squares Models and Related Chemometric Approaches in Metabonomics and Metabolic Phenotyping. J. Chemom. 2010, 24, 636–649.
- Noble, R.A.; Thomas, H.; Zhao, Y.; Herendi, L.; Howarth, R.; Dragoni, I.; Keun, H.C.; Vellano, C.P.; Marszalek, J.R.; Wedge, S.R. Simultaneous Targeting of Glycolysis and Oxidative Phosphorylation as a Therapeutic Strategy to Treat Diffuse Large B-Cell Lymphoma. Br. J. Cancer 2022, 127, 937–947. [Google Scholar] [CrossRef] [PubMed]Madsen, R.; Lundstedt, T.; Trygg, J. Chemometrics in Metabolomics—A Review in Human Disease Diagnosis. Anal. Chim. Acta 2010, 659, 23–33.
- Zhang, J.; Medina-Cleghorn, D.; Bernal-Mizrachi, L.; Bracci, P.M.; Hubbard, A.; Conde, L.; Riby, J.; Nomura, D.K.; Skibola, C.F. The Potential Relevance of the Endocannabinoid, 2-Arachidonoylglycerol, in Diffuse Large B-Cell Lymphoma. Oncoscience 2016, 3, 31–41. [Google Scholar] [CrossRef][Green Version]Broadhurst, D.I.; Kell, D.B. Statistical Strategies for Avoiding False Discoveries in Metabolomics and Related Experiments. Metabolomics 2007, 2, 171–196.
- Ducker, G.S.; Ghergurovich, J.M.; Mainolfi, N.; Suri, V.; Jeong, S.K.; Hsin-Jung Li, S.; Friedman, A.; Manfredi, M.G.; Gitai, Z.; Kim, H.; et al. Human SHMT Inhibitors Reveal Defective Glycine Import as a Targetable Metabolic Vulnerability of Diffuse Large B-Cell Lymphoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11404–11409. [Google Scholar] [CrossRef][Green Version]Blekherman, G.; Laubenbacher, R.; Cortes, D.F.; Mendes, P.; Torti, F.M.; Akman, S.; Torti, S.V.; Shulaev, V. Bioinformatics Tools for Cancer Metabolomics. Metabolomics 2011, 7, 329–343.
- Xiong, J.; Wang, L.; Fei, X.-C.; Jiang, X.-F.; Zheng, Z.; Zhao, Y.; Wang, C.-F.; Li, B.; Chen, S.-J.; Janin, A.; et al. MYC Is a Positive Regulator of Choline Metabolism and Impedes Mitophagy-Dependent Necroptosis in Diffuse Large B-Cell Lymphoma. Blood Cancer J. 2017, 7, e582. [Google Scholar] [CrossRef][Green Version]Barberini, L.; Noto, A.; Fattuoni, C.; Satta, G.; Zucca, M.; Cabras, M.G.; Mura, E.; Cocco, P. The Metabolomic Profile of Lymphoma Subtypes: A Pilot Study. Molecules 2019, 24, 13.
- Pera, B.; Krumsiek, J.; Assouline, S.E.; Marullo, R.; Patel, J.; Phillip, J.M.; Román, L.; Mann, K.K.; Cerchietti, L. Metabolomic Profiling Reveals Cellular Reprogramming of B-Cell Lymphoma by a Lysine Deacetylase Inhibitor through the Choline Pathway. EBioMedicine 2018, 28, 80–89. [Google Scholar] [CrossRef][Green Version]Schmidt, D.R.; Patel, R.; Kirsch, D.G.; Lewis, C.A.; Vander Heiden, M.G.; Locasale, J.W. Metabolomics in Cancer Research and Emerging Applications in Clinical Oncology. CA. Cancer J. Clin. 2021, 71, 333–358.
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. (Eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; WHO: Geneva, Switzerland, 2017. [Google Scholar]Pettersen, H.S.; Galashevskaya, A.; Doseth, B.; Sousa, M.M.L.; Sarno, A.; Visnes, T.; Aas, P.A.; Liabakk, N.-B.; Slupphaug, G.; Sætrom, P.; et al. AID Expression in B-Cell Lymphomas Causes Accumulation of Genomic Uracil and a Distinct AID Mutational Signature. DNA Repair 2015, 25, 60–71.
- Monrad, I.; Madsen, C.; Lauridsen, K.L.; Honoré, B.; Plesner, T.L.; Hamilton-Dutoit, S.; D’Amore, F.; Ludvigsen, M. Glycolytic Biomarkers Predict Transformation in Patients with Follicular Lymphoma. PLoS ONE 2020, 15, e0233449. [Google Scholar] [CrossRef]Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in b Cells. Cell Metab. 2012, 15, 110–121.
- Böttcher, M.; Baur, R.; Stoll, A.; Mackensen, A.; Mougiakakos, D. Linking Immunoevasion and Metabolic Reprogramming in B-Cell–Derived Lymphomas. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Banoei, M.M.; Mahé, E.; Mansoor, A.; Stewart, D.; Winston, B.W.; Habibi, H.R.; Shabani-Rad, M.-T. NMR-Based Metabolomic Profiling Can Differentiate Follicular Lymphoma from Benign Lymph Node Tissues and May Be Predictive of Outcome. Sci. Rep. 2022, 12, 8294. [Google Scholar] [CrossRef]
- Yi, S.; Zou, D.; Young, K.H. Decipher the 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Natl. Med. J. China 2016, 96, 3365–3369. [Google Scholar] [CrossRef]
- Sekihara, K.; Saitoh, K.; Han, L.; Ciurea, S.; Yamamoto, S.; Kikkawa, M.; Kazuno, S.; Taka, H.; Kaga, N.; Arai, H.; et al. Targeting Mantle Cell Lymphoma Metabolism and Survival through Simultaneous Blockade of MTOR and Nuclear Transporter Exportin-1. Oncotarget 2017, 8, 34552–34564. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hess, G.; Herbrecht, R.; Romaguera, J.; Verhoef, G.; Crump, M.; Gisselbrecht, C.; Laurell, A.; Offner, F.; Strahs, A.; Berkenblit, A.; et al. Phase III Study to Evaluate Temsirolimus Compared with Investigator’s Choice Therapy for the Treatment of Relapsed or Refractory Mantle Cell Lymphoma. J. Clin. Oncol. 2009, 27, 3822–3829. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the Role of MTOR in Cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef][Green Version]
- Lee, S.-C.; Shestov, A.A.; Guo, L.; Zhang, Q.; Roman, J.C.; Liu, X.; Wang, H.Y.; Pickup, S.; Nath, K.; Lu, P.; et al. Metabolic Detection of Bruton’s Tyrosine Kinase Inhibition in Mantle Cell Lymphoma Cells. Mol. Cancer Res. 2019, 17, 1365–1377. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reina-Campos, M.; Diaz-Meco, M.T.; Moscat, J. The Complexity of the Serine Glycine One-Carbon Pathway in Cancer. J. Cell Biol. 2020, 219, e201907022. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Du, J.; Zhang, H.; Ruan, G.; Xiang, J.; Wang, L.; Sun, H.; Guan, A.; Shen, G.; Liu, Y.; et al. Serum Metabolomics of Burkitt Lymphoma Mouse Models. PLoS ONE 2017, 12, e0170896. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Scarfò, L.; Ferreri, A.J.M.; Ghia, P. Chronic Lymphocytic Leukaemia. Crit. Rev. Oncol. Hematol. 2016, 104, 169–182. [Google Scholar] [CrossRef]
- La Vecchia, S.; Sebastián, C. Metabolic Pathways Regulating Colorectal Cancer Initiation and Progression. Semin. Cell Dev. Biol. 2020, 98, 63–70. [Google Scholar] [CrossRef]
- Rozovski, U.; Hazan-Halevy, I.; Barzilai, M.; Keating, M.J.; Estrov, Z. Metabolism Pathways in Chronic Lymphocytic Leukemia. Leuk. Lymphoma 2016, 57, 758–765. [Google Scholar] [CrossRef][Green Version]
- Rozovski, U.; Grgurevic, S.; Bueso-Ramos, C.; Harris, D.M.; Li, P.; Liu, Z.; Wu, J.Y.; Jain, P.; Wierda, W.; Burger, J.; et al. Aberrant LPL Expression, Driven by STAT3, Mediates Free Fatty Acid Metabolism in CLL Cells. Mol. Cancer Res. 2015, 13, 944–953. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Falchi, L.; Keating, M.J.; Marom, E.M.; Truong, M.T.; Schlette, E.J.; Sargent, R.L.; Trinh, L.; Wang, X.; Smith, S.C.; Jain, N.; et al. Correlation between FDG/PET, Histology, Characteristics, and Survival in 332 Patients with Chronic Lymphoid Leukemia. Blood 2014, 123, 2783–2790. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shalhout, S.; Haddad, D.; Sosin, A.; Holland, T.C.; Al-Katib, A.; Martin, A.; Bhagwat, A.S. Genomic Uracil Homeostasis during Normal B Cell Maturation and Loss of This Balance during B Cell Cancer Development. Mol. Cell. Biol. 2014, 34, 4019–4032. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tili, E.; Michaille, J.-J.; Luo, Z.; Volinia, S.; Rassenti, L.Z.; Kipps, T.J.; Croce, C.M. The Down-Regulation of MiR-125b in Chronic Lymphocytic Leukemias Leads to Metabolic Adaptation of Cells to a Transformed State. Blood 2012, 120, 2631–2638. [Google Scholar] [CrossRef][Green Version]
- Medriano, C.A.D.; Na, J.; Lim, K.-M.; Chung, J.-H.; Park, Y.H. Liquid Chromatography Mass Spectrometry-Based Metabolite Pathway Analyses of Myeloma and Non-Hodgkin’s Lymphoma Patients. Cell J. 2017, 19 (Suppl. 1), 44–54. [Google Scholar] [CrossRef]
- Han, J.; Li, Q.; Chen, Y.; Yang, Y. Recent Metabolomics Analysis in Tumor Metabolism Reprogramming. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef]
- MacIntyre, D.A.; Jiménez, B.; Lewintre, E.J.; Martín, C.R.; Schäfer, H.; Ballesteros, C.G.; Mayans, J.R.; Spraul, M.; García-Conde, J.; Pineda-Lucena, A. Serum Metabolome Analysis by 1H-NMR Reveals Differences between Chronic Lymphocytic Leukaemia Molecular Subgroups. Leukemia 2010, 24, 788–797. [Google Scholar] [CrossRef][Green Version]
- Alfaifi, A.; Bahashwan, S.; Alsaadi, M.; Malhan, H.; Aqeel, A.; Al-Kahiry, W.; Almehdar, H.; Qadri, I. Metabolic Biomarkers in B-Cell Lymphomas for Early Diagnosis and Prediction, as Well as Their Influence on Prognosis and Treatment. Diagnostics 2022, 12, 394. [Google Scholar] [CrossRef]
- Mamas, M.; Dunn, W.B.; Neyses, L.; Goodacre, R. The Role of Metabolites and Metabolomics in Clinically Applicable Biomarkers of Disease. Arch. Toxicol. 2011, 85, 5–17. [Google Scholar] [CrossRef]
- Beger, R.D. A Review of Applications of Metabolomics in Cancer. Metabolites 2013, 3, 552. [Google Scholar] [CrossRef][Green Version]
- Nalbantoglu, S. Metabolomics: Basic Principles and Strategies. In Molecular Medicine; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef][Green Version]
- Carneiro, G.; Radcenco, A.L.; Evaristo, J.; Monnerat, G. Novel Strategies for Clinical Investigation and Biomarker Discovery: A Guide to Applied Metabolomics. Horm. Mol. Biol. Clin. Investig. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.; Lopata, A.L.; Dasouki, M.; Abdel Rahman, A.M. Metabolomics toward Personalized Medicine. Mass Spectrom. Rev. 2019, 38, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Sévin, D.C.; Kuehne, A.; Zamboni, N.; Sauer, U. Biological Insights through Nontargeted Metabolomics. Curr. Opin. Biotechnol. 2015, 34, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mirnaghi, F.S.; Caudy, A.A. Challenges of Analyzing Different Classes of Metabolites by a Single Analytical Method. Bioanalysis 2014, 6, 3393–3416. [Google Scholar] [CrossRef]
- Spratlin, J.L.; Serkova, N.J.; Eckhardt, S.G. Clinical Applications of Metabolomics in Oncology: A Review. Clin. Cancer Res. 2009, 15, 431–440. [Google Scholar] [CrossRef][Green Version]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for Large-Scale Metabolic Profiling of Serum and Plasma Using Gas Chromatography and Liquid Chromatography Coupled to Mass Spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Ganti, S.; Weiss, R.H. Urine Metabolomics for Kidney Cancer Detection and Biomarker Discovery. Urol. Oncol. Semin. Orig. Investig. 2011, 29, 551–557. [Google Scholar] [CrossRef][Green Version]
- Nielsen, T.H.; Diaz, Z.; Christodoulopoulos, R.; Charbonneau, F.; Qureshi, S.; Rousseau, C.; Benlimame, N.; Camlioglu, E.; Constantin, A.M.; Oros, K.K.; et al. Methods for Sample Acquisition and Processing of Serial Blood and Tumor Biopsies for Multicenter Diffuse Large B-Cell Lymphoma Clinical Trials. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 2688–2693. [Google Scholar] [CrossRef][Green Version]
- Larkin, J.R.; Anthony, S.; Johanssen, V.A.; Yeo, T.; Sealey, M.; Yates, A.G.; Smith, C.F.; Claridge, T.D.W.; Nicholson, B.D.; Moreland, J.-A.; et al. Metabolomic Biomarkers in Blood Samples Identify Cancers in a Mixed Population of Patients with Nonspecific Symptoms. Clin. Cancer Res. 2022, 28, 1651–1661. [Google Scholar] [CrossRef]
- Stenson, M. Diffuse Large B-Cell Lymphoma—Proteomic and Metabolomic Studies on Prognosis and Treatment Failure. Ph.D. Thesis, Gothenburg University, Gothenburg, Sweden, 2018. [Google Scholar]
- González-Domínguez, R.; González-Domínguez, Á.; Sayago, A.; Fernández-Recamales, Á. Recommendations and Best Practices for Standardizing the Pre-Analytical Processing of Blood and Urine Samples in Metabolomics. Metabolites 2020, 10, 229. [Google Scholar] [CrossRef]
- Rochat, B.; Mohamed, R.; Sottas, P.-E. LC-HRMS Metabolomics for Untargeted Diagnostic Screening in Clinical Laboratories: A Feasibility Study. Metabolites 2018, 8, 39. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bujak, R.; Struck-Lewicka, W.; Markuszewski, M.J.; Kaliszan, R. Metabolomics for Laboratory Diagnostics. J. Pharm. Biomed. Anal. 2015, 113, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Stevens, V.L.; Hoover, E.; Wang, Y.; Zanetti, K.A. Pre-Analytical Factors That Affect Metabolite Stability in Human Urine, Plasma, and Serum: A Review. Metabolites 2019, 9, 156. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nicholson, J.K.; Holmes, E.; Kinross, J.M.; Darzi, A.W.; Takats, Z.; Lindon, J.C. Metabolic Phenotyping in Clinical and Surgical Environments. Nature 2012, 491, 384–392. [Google Scholar] [CrossRef]
- Pitt, J.J. Principles and Applications of Liquid Chromatography-Mass Spectrometry in Clinical Biochemistry. Clin. Biochem. Rev. 2009, 30, 19–34. [Google Scholar]
- Kuehnbaum, N.L.; Britz-McKibbin, P. New Advances in Separation Science for Metabolomics: Resolving Chemical Diversity in a Post-Genomic Era. Chem. Rev. 2013, 113, 2437–2468. [Google Scholar] [CrossRef]
- Li, L.-H.; Hsieh, H.-Y.; Hsu, C.-C. Clinical Application of Ambient Ionization Mass Spectrometry. Mass Spectrom. 2017, 6, S0060. [Google Scholar] [CrossRef][Green Version]
- Zhou, B.; Xiao, J.F.; Tuli, L.; Ressom, H.W. LC-MS-Based Metabolomics. Mol. BioSyst. 2012, 8, 470–481. [Google Scholar] [CrossRef][Green Version]
- Fiehn, O. Metabolomics by Gas Chromatography–Mass Spectrometry: Combined Targeted and Untargeted Profiling. Curr. Protoc. Mol. Biol. 2016, 114. [Google Scholar] [CrossRef][Green Version]
- Koek, M.M.; Jellema, R.H.; van der Greef, J.; Tas, A.C.; Hankemeier, T. Quantitative Metabolomics Based on Gas Chromatography Mass Spectrometry: Status and Perspectives. Metabolomics 2011, 7, 307–328. [Google Scholar] [CrossRef][Green Version]
- Bueno Duarte, G.H.; de Piloto Fernandes, A.M.A.; Silva, A.A.R.; Zamora-Obando, H.R.; Amaral, A.G.; de Sousa Mesquita, A.; Schmidt-Filho, J.; Cordeiro de Lima, V.C.; D’Almeida Costa, F.; Andrade, V.P.; et al. Gas Chromatography-Mass Spectrometry Untargeted Profiling of Non-Hodgkin’s Lymphoma Urinary Metabolite Markers. Anal. Bioanal. Chem. 2020, 412, 7469–7480. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.N.; Fan, T.W.-M. NMR-Based Stable Isotope Resolved Metabolomics in Systems Biochemistry. Arch. Biochem. Biophys. 2017, 628, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.W.-M.; Lane, A.N. Applications of NMR Spectroscopy to Systems Biochemistry. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 92–93, 18–53. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nagana Gowda, G.A.; Raftery, D. Can NMR Solve Some Significant Challenges in Metabolomics? J. Magn. Reson. 2015, 260, 144–160. [Google Scholar] [CrossRef][Green Version]
- Tiziani, S.; Kang, Y.; Choi, J.S.; Roberts, W.; Paternostro, G. Metabolomic High-Content Nuclear Magnetic Resonance-Based Drug Screening of a Kinase Inhibitor Library. Nat. Commun. 2011, 2, 545. [Google Scholar] [CrossRef][Green Version]
- Dunn, W.B.; Wilson, I.D.; Nicholls, A.W.; Broadhurst, D. The Importance of Experimental Design and QC Samples in Large-Scale and MS-Driven Untargeted Metabolomic Studies of Humans. Bioanalysis 2012, 4, 2249–2264. [Google Scholar] [CrossRef][Green Version]
- Fonville, J.M.; Richards, S.E.; Barton, R.H.; Boulange, C.L.; Ebbels, T.M.D.; Nicholson, J.K.; Holmes, E.; Dumas, M.-E. The Evolution of Partial Least Squares Models and Related Chemometric Approaches in Metabonomics and Metabolic Phenotyping. J. Chemom. 2010, 24, 636–649. [Google Scholar] [CrossRef]
- Madsen, R.; Lundstedt, T.; Trygg, J. Chemometrics in Metabolomics—A Review in Human Disease Diagnosis. Anal. Chim. Acta 2010, 659, 23–33. [Google Scholar] [CrossRef]
- Broadhurst, D.I.; Kell, D.B. Statistical Strategies for Avoiding False Discoveries in Metabolomics and Related Experiments. Metabolomics 2007, 2, 171–196. [Google Scholar] [CrossRef][Green Version]
- Blekherman, G.; Laubenbacher, R.; Cortes, D.F.; Mendes, P.; Torti, F.M.; Akman, S.; Torti, S.V.; Shulaev, V. Bioinformatics Tools for Cancer Metabolomics. Metabolomics 2011, 7, 329–343. [Google Scholar] [CrossRef][Green Version]
- Wishart, D.S. Emerging Applications of Metabolomics in Drug Discovery and Precision Medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Ayat, M. 3-Bromopyruvate as a Promising Treatment for Hematological Cancer. J. Cancer Res. Treat. 2018, 6, 12–17. [Google Scholar] [CrossRef][Green Version]
- Zhou, L.; Ding, L.; Gong, Y.; Zhao, J.; Zhang, J.; Mao, Z.; Wang, Z.; Zhang, W.; Zhou, R. NEK2 Promotes Cell Proliferation and Glycolysis by Regulating PKM2 Abundance via Phosphorylation in Diffuse Large B-Cell Lymphoma. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.-F.; Yu, Q.-Q.; Young, K.H. Critically Dysregulated Signaling Pathways and Clinical Utility of the Pathway Biomarkers in Lymphoid Malignancies. Chronic Dis. Transl. Med. 2018, 4, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, M.; Peperzak, V. Advances and Perspectives in the Treatment of B-Cell Malignancies. Cancers 2021, 13, 2266. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.E.; Chiche, J. Metabolic Reprogramming of Non-Hodgkin’s B-Cell Lymphomas and Potential Therapeutic Strategies. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Adekola, K.U.A.; Dalva Aydemir, S.; Ma, S.; Zhou, Z.; Rosen, S.T.; Shanmugam, M. Investigating and Targeting Chronic Lymphocytic Leukemia Metabolism with the Human Immunodeficiency Virus Protease Inhibitor Ritonavir and Metformin. Leuk. Lymphoma 2015, 56, 450–459. [Google Scholar] [CrossRef][Green Version]
- Brown, J.R.; Byrd, J.C.; Coutre, S.E.; Benson, D.M.; Flinn, I.W.; Wagner-Johnston, N.D.; Spurgeon, S.E.; Kahl, B.S.; Bello, C.; Webb, H.K.; et al. Idelalisib, an Inhibitor of Phosphatidylinositol 3-Kinase P110δ, for Relapsed/Refractory Chronic Lymphocytic Leukemia. Blood 2014, 123, 3390–3397. [Google Scholar] [CrossRef]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kδ Inhibition by Idelalisib in Patients with Relapsed Indolent Lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef][Green Version]
- Galicia-Vázquez, G.; Smith, S.; Aloyz, R. Del11q-Positive CLL Lymphocytes Exhibit Altered Glutamine Metabolism and Differential Response to GLS1 and Glucose Metabolism Inhibition. Blood Cancer J. 2018, 8, 13. [Google Scholar] [CrossRef][Green Version]
- Ruella, M.; Kenderian, S.S.; Shestova, O.; Fraietta, J.A.; Qayyum, S.; Zhang, Q.; Maus, M.V.; Liu, X.; Nunez-Cruz, S.; Klichinsky, M.; et al. The Addition of the BTK Inhibitor Ibrutinib to Anti-CD19 Chimeric Antigen Receptor T Cells (CART19) Improves Responses against Mantle Cell Lymphoma. Clin. Cancer Res. 2016, 22, 2684–2696. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Noble, R.A.; Bell, N.; Blair, H.; Sikka, A.; Thomas, H.; Phillips, N.; Nakjang, S.; Miwa, S.; Crossland, R.; Rand, V.; et al. Inhibition of Monocarboxyate Transporter 1 by AZD3965 as a Novel Therapeutic Approach for Diffuse Large B-Cell Lymphoma and Burkitt Lymphoma. Haematologica 2017, 102, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.C.; Kong, S.-Y.; Jang, S.-G.; Kim, K.-H.; Ahn, S.-A.; Park, W.-S.; Park, S.; Yun, T.; Eom, H.-S. Identification of Hypoxanthine as a Urine Marker for Non-Hodgkin Lymphoma by Low-Mass-Ion Profiling. BMC Cancer 2010, 10, 55. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kirsch, B.J.; Chang, S.J.; Betenbaugh, M.J.; Le, A. Non-Hodgkin Lymphoma Metabolism. Adv. Exp. Med. Biol. 2021, 1311, 103–116. [Google Scholar] [CrossRef]
- Pettersen, H.S.; Galashevskaya, A.; Doseth, B.; Sousa, M.M.L.; Sarno, A.; Visnes, T.; Aas, P.A.; Liabakk, N.-B.; Slupphaug, G.; Sætrom, P.; et al. AID Expression in B-Cell Lymphomas Causes Accumulation of Genomic Uracil and a Distinct AID Mutational Signature. DNA Repair 2015, 25, 60–71. [Google Scholar] [CrossRef]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in b Cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef][Green Version]