Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Folkert Kuipers and Version 2 by Camila Xu.
Bile acids represent a class of cholesterol derivatives that is essential for intestinal absorption of lipids and fat-soluble vitamins, plays an important role in cholesterol turnover and, as more recently discovered, acts as a group of signaling molecules that exerts hormonal functions throughout the body.
- gut microbiota
- bile acids
- cardiovascular disease
- atherosclerosis
1. Bile Acids Are Synthesized by the Liver and Extensively Metabolized by the Microbiota
Bile acids (BAs) are amphipathic cholesterol derivates, characterized by one or more hydroxyl groups at their steroid nucleus (in mammals, commonly at positions 3, 7, and/or 12) and a shortened side chain bearing a carboxylic acid group [1][22] (Figure 1A). BAs are exclusively synthesized in hepatocytes in a process that involves a cascade of enzymatic steps in different compartments of the cell [2][3][4][23,24,25]. The products of this multi-step process are the so-called primary BAs, i.e., cholic acid (CA) and chenodeoxycholic acid (CDCA) in humans, which are products of the well-established “classical” or “alternative” pathways [5][26].
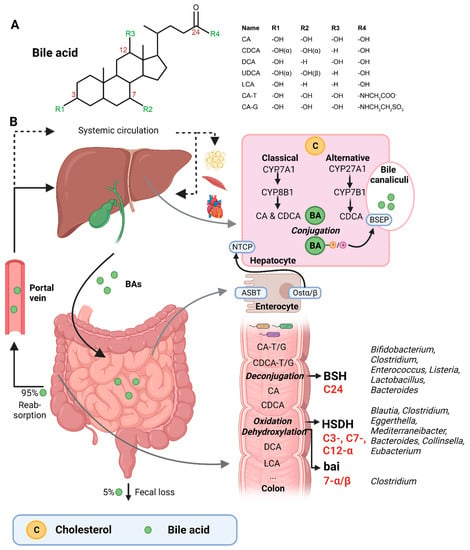
Figure 1. (A) Structural formula of bile acid. (B) Hepatic BA synthesis, enterohepatic circulation, and microbial BA modifications in the human body. BA = bile acid; CA = cholic acid; CDCA = chenodeoxycholic acid; DCA = deoxycholic acid; UDCA = ursodeoxycholic acid; LCA = lithocholic acid; TCA = taurocholic acid; GCA = glycocholic acid; C = cholesterol; BSEP = bile salt-export pump; NTCP = Na+-taurocholic acid co-transporting polypeptide; ASBT = apical sodium-dependent BA transporter; Ostα/β = organic solute transporter α/β; BSH = bile salt hydrolases; HSDH = hydroxysteroid dehydrogenase; bai = BA inducible genes. This illustration was created with Biorender.com (accessed on 1 February 2023).
The first step of the classical pathway is catalyzed by the rate-controlling enzyme cytochrome P450 cholesterol 7α-hydroxylase (CYP7A1), which yields 7α-hydroxycholesterol [2][6][23,27] (Figure 1B). The alternative pathway is initiated by sterol 27α-hydroxylase (CYP27A1), and further processed by oxysterol 7α-hydrolase (CYP7B1) [6][27]. The 7α-hydroxylated intermediates from both the classical and alternative pathway undergo several sterol ring modifications, as well as side chain oxidation and shortening, leading to the production of CDCA [5][6][26,27]. In addition, the classical pathway also generates CA, which involves the activity of sterol 12α-hydroxylase (CYP8B1) [7][28]. Of note, rodents produce additional forms of primary BAs in the liver—α- and β muricholic acids (MCAs) and ursodeoxycholic acid (UDCA)—which are derived from CDCA by mouse/rat-specific cytochrome P450 2C70 (Cyp2c70) [5][8][26,29].
Primary BAs are conjugated with either taurine or glycine at the C24 position, which allows for their active secretion into the bile and decreases their pKa, and hence prevents passive reabsorption in the upper intestine [4][25]. BAs are secreted into the bile canaliculi by the bile salt-export pump (BSEP) and stored in the gallbladder in high mM range [5][9][26,30] (Figure 1B).
Upon meal intake, cephalic-phase-induced cholecystokinin secretion from the duodenum stimulates gallbladder contraction, leading to the discharge of stored BAs into the intestine [4][25]. In the intestinal lumen, BAs solubilize lipids, including cholesterol, and fat-soluble vitamins. Most primary BAs are actively reabsorbed in the ileum by the apical sodium-dependent BA transporter (ASBT) [4][25]. A small proportion of BAs escape uptake and reach the colon, where the gut microbiota can convert primary into secondary BAs (lithocholic acid (LCA) and deoxycholic acid (DCA)), a process that involves three major groups of bacterial enzymes (discussed in the subsequent section) [3][24]. Unconjugated secondary BAs are relatively hydrophobic and can thus be passively reabsorbed from the colon [10][20]. Most BAs (95%) are reabsorbed in the ileum or colon, and via the portal vein they are delivered back to the liver, whereafter they are taken up by Na+-taurocholic acid co-transporting polypeptide (NTCP), in order to finalize a process called the enterohepatic circulation. In the liver, unconjugated primary and secondary BAs are re-conjugated and secreted into bile. Only a minor amount (~5%) of BAs, which are not taken up in the ileum or colon, are excreted in feces per cycle [10][20]. This loss of BAs is compensated by the de novo synthesis of BAs in the liver to maintain the size of the circulating BA pool [10][20].
A relatively small fraction of BAs escape first pass clearance by the liver and flow into the systemic circulation, with concentrations in the µM range [9][30]. From the peripheral circulation, BAs can reach multiple organs, including adipose tissue, muscle, and the heart [11][12][31,32]. BAs exert hormone-like functions by acting as signaling molecules and activate several BARs, i.e., nuclear receptors—farnesoid X receptor (FXR), vitamin D receptor (VDR), pregnane R receptor (PXR)—and membrane-bound receptors—Takeda G protein-coupled receptor (TGR5) and muscarinic receptors (MRs). Through these actions, BAs regulate their own homeostasis, as well as lipid and glucose metabolism, intestinal barrier function, cardiovascular functions, and inflammation [11][13][14][21,31,33]. Importantly, different BA species have dissimilar affinities for the activation of BARs (FXR: CDCA > LCA = DCA > CA; TGR5: LCA > DCA > CDCA > CA; VDR: LCA; PXR: LCA = CDCA = DCA = CA) [9][15][30,34]. Differences in BA pool composition, which are prominent across individuals [16][35], will thus affect BA signaling in a personalized manner.
2. Bacteria Involved in Bile Acid Metabolism
The three major groups of bacterial enzymes—bile salt hydrolases (BSHs), hydroxysteroid dehydrogenases (HSDHs), and bile-acid-inducible (bai) genes—are responsible for the generation of secondary BAs in the colon, leading to a major increase of the diversity of the BA pool. The major structural modifications include deconjugation, which is an obligatory first step; oxidation of hydroxy groups at the C3, C7, and C12 position; and 7α/β-dehydroxylation [3][4][24,25]. In addition, (7α/β, 3α/β) isomerization- and (5-H α/β) epimerization modifications give rise to UDCA and iso- and allo-BAs, respectively [5][26].
BSHs catalyze the deconjugation of the N-acylamide bond between primary BAs and taurine or glycine at the C24 position [4][25]. BSHs have been identified in several microbial genera, including Bifidobacterium [9][17][30,36], Clostridium [9][18][30,37], Enterococcus [9][30], Listeria [9][19][20][30,38,39], Lactobacillus [9][21][22][30,40,41], and Bacteroides [23][42]. Recently, computational analyses have shown that the human gut microbiota contains 591 intestinal bacterial strains within 117 genera with BSHs sequences [24][43]. BSHs, encompass seven [25][44] or eight sub-groups [24][43], showing differences in deconjugation ability. BSH-T3 shows the highest enzyme and deconjugation activity and is only found in Lactobacillus [24][43]. Recently, research has shown that, after deconjugation, the gut bacteria can also mediate the conjugation of the CA backbone with the amino acids phenylalanine, tyrosine, or leucine [26][45]. The microbial enzyme responsible for these BA modifications remains unknown. Interestingly, these amino acid BA conjugates are found in humans and are enriched in patients with inflammatory bowel disease or cystic fibrosis [26][45].
The second major group of bacterial enzymes are HSDHs, which oxidize and epimerize C3, C7, and C12 hydroxy groups of BAs. HSDH enzymes have been identified in the microbial genera Blautia (3α), Clostridium (3-, 7-, 12α), Eggerthella (3-, 12α), Mediterraneibacter (3α), Bacteroides (7α), Collinsella (7α), and Eubacterium (7α) [9][30]. Epimerization of hydroxy groups leads to a reversible change from the α to the β configuration, generating stable oxo-BAs as intermediates [4][25]. The reaction depends, in part, on the redox potential of the environment. For example, oxo-BAs are more present at the mucosal surface, where the redox potential is high, whereas less oxo-BAs are present in the lumen of the large intestine, where the redox potential is low [3][24]. Interestingly, the production of 12-oxoCDCA may reduce the formation of secondary BA DCA [27][46], which has been implicated in liver [28][47] and colon cancer [29][48], cholesterol gallstone formation [30][49], and CVD [31][50].
Bacteria that carry the bai operon produce enzymes that carry out 7-α/β dihydroxylation, resulting in the major secondary BAs DCA and LCA. Surprisingly, this metabolic pathway is only found in 0.0001% of colonic gut microbiota, belonging to the genera Clostridium [3][4][32][24,25,51]. Moreover, 7-α/β dehydroxylation only takes place after deconjugation, implicating a functional interplay between deconjugation and dehydroxylation [3][24].
Thus, the gut microbiota is responsible for diversifying the BA pool. This strongly affects BA signaling, as BAs have different affinities towards BARs [10][20]. Importantly, the regulation between the gut microbiota and BAs is reciprocal, meaning that BAs can also modulate the gut microbiota either by direct or indirect effects. For example, BAs can disrupt bacterial membranes or bind to intestinal FXR, promoting the expression of antimicrobial agents [33][52]. Moreover, conjugated BAs play an important role in the prevention of bacterial overgrowth in the proximal small intestine, which is relatively devoid of microbes under normal conditions [34][53]. Studies have shown that replenishing intestinal BA concentrations in BA-deficient rats abolished bacterial overgrowth in the small intestine [34][53]. On the other hand, bacteria that inhabit the intestinal tract must have specific resistance mechanisms to protect themselves against bile [35][54]. For example, Lactobacillus and Bifidobacterium produce proteins that are devoted to the efflux of BAs [35][54].
Of note, gut bacteria can also directly metabolize cholesterol in the intestine via dehydrogenase activity encoded by intestinal sterol metabolism A (ismA) genes, producing cholestanone and coprostanol [36][55]. These genes are found in the human gut microbiota in geographically diverse human cohorts and show a negative correlation with circulating cholesterol levels [36][55]. In addition, recent studies highlight the bacterial potential to sulfonate cholesterol and related steroids in the gut. The sulfotransferase enzyme is identified in the microbial species Bacteroides thetaiotaomicron [37][38][56,57]. The new (direct) lipid-metabolizing functions of the gut microbiota, which was originally thought to be performed by host enzymes, represent a great breakthrough in the understanding of cholesterol homeostasis.
3. Gut Microbiota Signatures in Cardiovascular Disease
Since the late 1980s, researchers have implicated a role of bacteria in atherogenesis [39][58]. In these early studies, the bacterium Chlamydia pneumoniae was associated with CAD and myocardial infarction. A few years later, bacterial DNA from many other bacteria genera/species were found in human atherosclerotic plaques [40][41][42][59,60,61], whereas healthy tissue (e.g., non-transplanted hearts) does not contain bacterial DNA [43][62]. Bacterial DNA has also been linked to inflammation, as the amount of bacterial DNA was found to correlate with the number of leukocytes in the plaque [40][59]. This suggests that the underlying pathophysiology of atherosclerosis may involve bacterial activation of the immune system. In addition, bacteria originating from the gut and the oral cavity matched with bacterial DNA present in atherosclerotic plaques and correlated with disease biomarkers [40][59]. Thus, the re-allocation of bacteria from the intestinal tract to the heart may contribute to disease development, which has sparked interest to evaluate the role of the gut microbiota in CVD. In general, gut microbiota diversity has been found to negatively correlate with risk factors of atherosclerosis, such as obesity, hyperinsulinemia, hypertension, and dyslipidemia [44][45][46][72,80,81]. However, others have demonstrated that alpha diversity was either not different or increased in atherosclerosis patients compared with the controls [47][48][49][50][51][63,66,67,69,71]. These discrepancies could be due to limitations in the respective studies, such as omitting important confounders in their analyses (e.g., age, sex, and BMI) or assessing diversity with an estimation-based method (Chao1) [47][63]. Moreover, the temporal dynamics and intra- and interindividual heterogeneity of the gut microbiota underscores the difficulty in studying and comparing cross-sectional studies. Rodent models (especially germ-free models), which are more experimentally controlled, represent an invaluable tool for studying microbe–host interactions in the context of CVD.4. Altered Bile Acid Metabolism in Cardiovascular Disease
Examining the composition of the gut microbiota at one single point in time ignores the complex nature of the gut microbiota as an ecosystem. Investigating functional shifts of the gut microbiota, including changes in gut-derived metabolites, helps to identify measurable read-outs of bacterial functions in health and disease. BAs might be a relevant gut metabolite in relation to CVD, despite controversy across studies [52][53][54][55][86,87,88,89]. Lower plasma BA levels have been reported in CVD [52][53][86,87]. Low serum BAs appeared to be independently associated with the presence and severity of CAD, especially for the presence of myocardial infarction (MI) [53][87]. These results were largely confirmed by Nguyen et al., although serum BA concentrations in both the control and CAD patients were lower compared with the previous study [52][86]. The latter study also observed doubled serum BAs in patients receiving statin therapy, suggesting that serum BAs levels are amendable by statin administration in CAD patients. Statins are a commonly described drug to lower cholesterol via the inhibition of β-Hydroxy β-methylglutaryl-CoA (HMG-CoA) reductase, the rate limiting enzyme in the cholesterol biosynthesis pathway [56][90]. Moreover, glyco-CDCA was two-fold higher in CAD patients [52][86] and, together with the total serum BAs, were predictors of CAD [52][53][86,87]. In contrast, a previous study comparing CAD and non-CAD patients did not demonstrate a significant association between serum BAs and CAD [54][88]. In addition, the total serum BAs are known to increase in patients with liver cirrhosis, which is associated with cardiac dysfunction [55][89]; total serum BAs are elevated up to 100 times the normal values in patients with cirrhotic cardiomyopathy [55][89], i.e., much more compared with CAD patients. Although inconsistency is found between studies, it appears that either low or extreme high serum BAs can be associated with CVD. For zooming in on small-molecule metabolites in relation to CVD, untargeted metabolomics is a powerful tool to discover novel metabolites. Zhang et al. discovered that six metabolites were significantly altered in CAD patients [57][91]. LCA, together with 4-pyridocix acid and phosphatidylglycerol (20:3/2:0), showed the strongest positive correlation with CAD, defined as >80% stenosis in at least one artery. Of note, Chen et al. observed large inter-individual variability in plasma BA profiles in human obesity [16][35]. This variability suggests a more personalized approach to finding biomarkers and future therapeutic applications of BAs in CVD, although participants with recent cardiovascular events were excluded from this study [16][35]. Nevertheless, secondary BAs, i.e., DCA and LCA, were associated with diabetes and liver fat content, which are two risk factors of CVD, in these obese subjects. In addition to plasma BAs, fecal bile acid excretion (BAE), which equals hepatic BA synthesis under steady state conditions, has also been associated with CAD [58][59][60][92,93,94]. CAD-patients were found to excrete less BAs, particularly DCA and LCA, compared with non-CAD controls [58][59][60][92,93,94]. A historical follow-up of 20 years showed that BAE was a significant independent parameter that predicted CAD in humans, in which BAE < 415 mg/day was associated with a higher long-term mortality due to CAD [60][94]. More specifically, 75% of the patients with BAE < 262.4/day developed a stroke relative to none of the patients with BAE > 622 mg/day [59][93]. BAE can thus serve as an interesting biomarker of CAD. Thus, these studies provide evidence that measuring plasma and fecal BAs may aid in the assessment of the gut microbiota contributions to CVD. Understanding determinants of BA pool/metabolism and its reflection in CVD is important to rationalize their use as potential biomarkers and therapeutic targets.5. Bile Acids as Mediators of Cardiovascular Disease Risk
In this section, the focus switches from association to causality regarding the potential roles of BAs in CVD. The multifaceted roles of BAs in lipid homeostasis, immunity, and heart function indicate the ability to mediate CVD, as discussed in the following sections (Figure 2).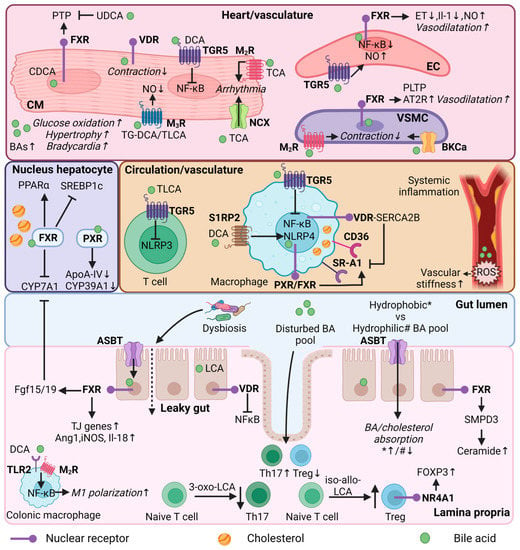
Figure 2. Bile acid regulation in lipid and immune metabolism, and heart function. Mechanistic effects of BAs in different organs (heart/vasculature/circulation/colon/lamina propria) or cell types (cardiomyocyte, endothelial cell, vascular smooth muscle cell, immune cells, enterocyte) in relation to lipid and immune metabolism, and heart function. CM = cardiomyocyte; EC = endothelial cell; VSMC = vascular smooth muscle cell; Th17—T helper 17 cells; Treg = Regulatory T cells; FXR = farnesoid X receptor; VDR = vitamin D receptor; PXR = pregnane R receptor; NR4A1 = nuclear receptor 4A1; TGR5 = Takeda G protein-coupled receptor; MR = muscarinic receptor; S1RP2 = sphingosine 1-phosphate receptor 2; SR-A1 = Class A1 scavenger receptors; AT2R = angiotensin II receptor type 2; TLR2—toll-like receptor 2; NCX = Na+/Ca2+ exchange protein; BKCa = large conductance Ca2+-activated K+; ASBT = apical sodium-dependent bile acid transporter; BA = bile acid; UDCA = ursodeoxycholic acid; (T/G)DCA = (tauro/glycol)deoxycholic acid; (T)LCA = Taurolithocholic acid; TCA = Taurocholic acid; CDCA = chenodeoxycholic acid; NF-κB = nuclear factor kappa B; PTP = Mitochondrial permeability transition pore; PLTP = phospholipid transfer protein; ET = endothelin-1; NO = nitric oxide; ROS = reactive oxygen species; PPARα = peroxisome proliferator-activated receptor alpha; SREBP1c = sterol regulatory element binding protein 1c; CYP7A1 = cytochrome P450 7A1; CYP39A1 = cytochrome P450 39A1; ApoA-IV = Apolipoprotein A-IV; NLRP3/4 = NLR family pyrin; FOXP3 = forkhead box P3; SMPD3 = Sphingomyelin Phosphodiesterase 3; SERCA2B = Sarcoendoplasmic reticulum calcium ATPase 2b; FGF15/19 = fibroblast growth factor 15/19; Ang1 = angiopoietin 1; iNOS = nitric oxide synthase; IL = interleukin; TJ = tight junction; * = hydrophobic bile acid pool; # = hydrophilic bile acid pool. This illustration was created with Biorender.com.