Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 1 by Zhiwen Wang.
Hemes belong to a small subgroup of the tetrapyrrole family, which is characterized by the combination of a ferrous ion and a porphyrin macrocycle. The ‘true’ hemes possess a fully oxidized porphyrin macrocycle, including heme a, heme b, heme c, and heme o. Hemes are an important class of prosthetic molecules that play roles in a number of biological processes. The most widespread and ubiquitous is heme b, which plays an important role in transporting oxygen as part of hemoglobin. In addition, heme b is a cofactor for many enzymes, such as myoglobin, cytochrome P450, and peroxidases, and plays significant roles in catalysis, transcription, signaling, and electron transfer.
- heme b
- pathway
- protein
1. Introduction
The capacity to synthesize heme b is very common but not ubiquitous, and there are organisms with a complete absence of a tetrapyrrole biosynthetic capacity or incomplete pathways. The available research suggests that the overall biosynthetic pathway of heme b can be divided into three parts (Figure 1): (i) the synthesis of the precursor 5-aminolevulinate (5-ALA) via the C4 or C5 pathway, (ii) the synthesis of the intermediate metabolite uroporphyrinogen III (UPG III) via a conserved core pathway, and (iii) the synthesis of heme b via the protoporphyrin-dependent (PPD) pathway, coproporphyrin-dependent (CPD) pathway, or siroheme-dependent (SHD) pathway.
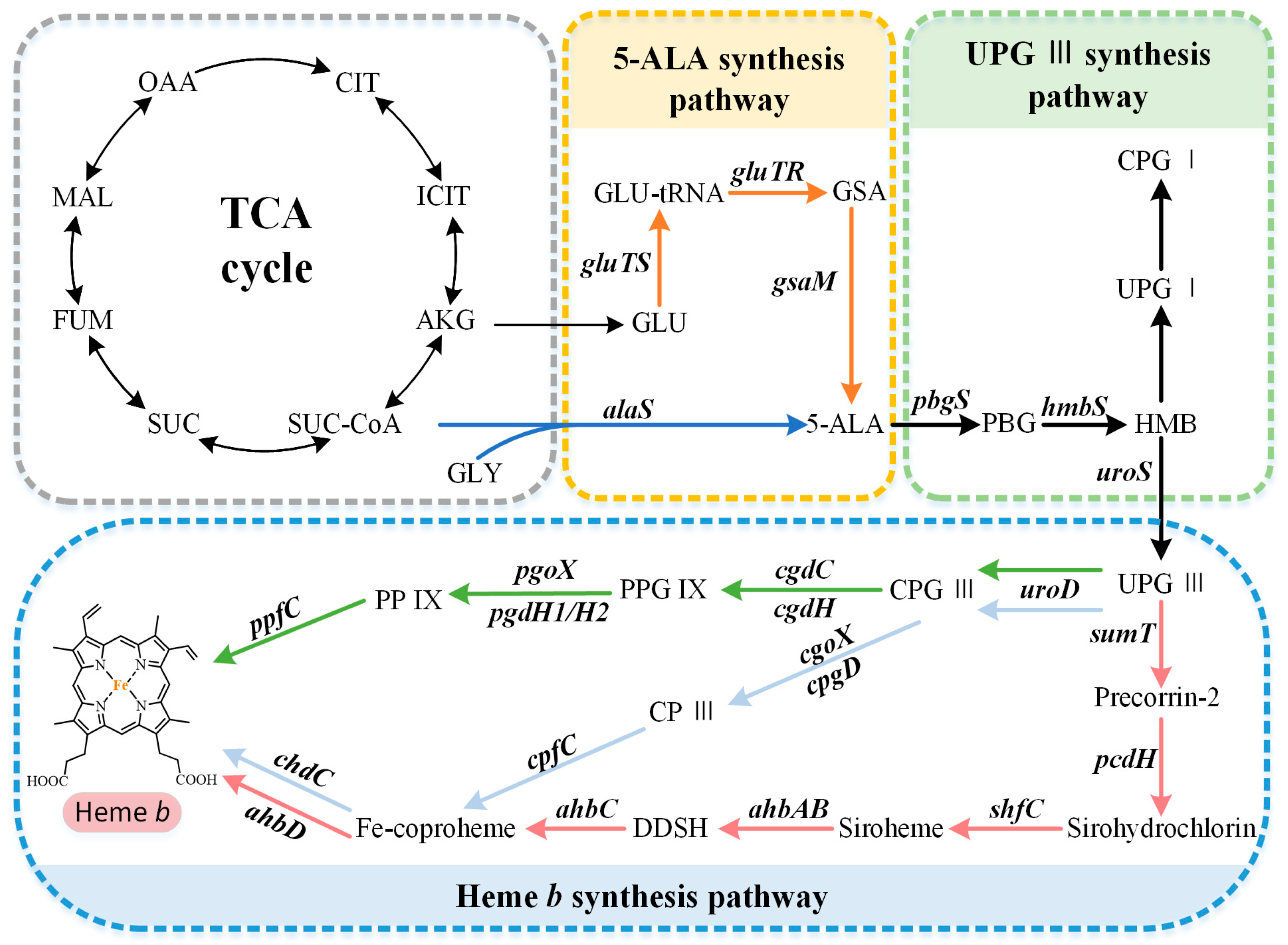
Figure 1. Heme b biosynthesis pathway. The overall biosynthetic pathway of heme b can be divided into three parts. The orange arrows indicate the C5 pathway, the blue arrows indicate the C4 pathway, the green arrows indicate the protoporphyrin-dependent (PPD) pathway, the light blue arrows indicate the coproporphyrin-dependent (CPD) pathway, and the red arrows indicate the siroheme-dependent (SHD) pathway. OAA, oxaloacetate; MAL, malate; FUM, fumarate; SUC, succinate; SUC-COA, succinyl-CoA; α-KG, α-oxoglutarate; GLY, glycine; GLU, glutamate; GLU-tRNA, glutamyl-tRNA; GSA, glutamate-1-semialdehyde; 5-ALA, 5-aminolevulinic acid; PBG, porphobilinogen; HMB, hydroxymethylbilane; UPG I, uroporphyrinogen I; CPG I, coproporphyrinogen I; UPG III, uroporphyrinogen III; CPG III, coproporphyrinogen III; PPG IX, protoporphyrinogen IX; PP IX, protoporphyrin IX; CP III, coproporphyrin III; DDSH, 12,18-didecarboxysiroheme.
2. Biosynthetic Pathways of the Precursor 5-ALA
The common precursor of tetrapyrroles, 5-ALA, is synthesized via two completely different biosynthetic pathways. The C4 pathway (Shemin pathway) was the first to be discovered, using succinyl-CoA and glycine as the substrates (Figure 1). It is mainly found in metazoans, fungi, and alphaproteobacteria [11][1]. For a long time, this was thought to be the only pathway for 5-ALA biosynthesis. However, in 1973, Beale et al. confirmed the existence of an alternative 5-ALA biosynthesis pathway, called the C5 pathway (Beale pathway), through their studies on cucumber cotyledons [15][2]. This route, which uses glutamate as the initial substrate, is predominantly found in plants, archaea, and most bacteria [11][1]. Generally, only one of these pathways is present in each organism, but a small number of organisms contain both pathways, such as Euglena gracilis [16][3].
The C4 pathway requires only one step to synthesize 5-ALA, which is catalyzed to the pyridoxal-5′-phosphate (PLP)-dependent ALA synthase (AlaS). This is a homodimeric protein, containing an active-site lysine that is covalently bound to the PLP cofactor, which catalyzes the condensation of succinyl-CoA and glycine, releasing carbon dioxide and coenzyme A in the process [16][3]. As shown in Figure 1, the C5 pathway is more complex than the C4 pathway, requiring three steps to synthesize 5-ALA from glutamate. First, glutamate is converted into Glu-tRNA in the presence of glutamyl-tRNA synthetase (GluTS). The next step is catalyzed using the NADPH-dependent glutamyl-tRNA reductase (GluTR). Its highly conserved cysteine residues bind to glutamyl-tRNA and release GlutRNA to form a thioester intermediate, which is then reduced to glutamate-1-semialdehyde (GSA) [17][4]. Finally, glutamate-1-semialdehyde-2,1-aminomutase (GsaM) catalyzes an intramolecular amine transfer, resulting in the conversion of GSA to 5-ALA [18][5]. GsaM also requires PLP as a cofactor and has a structural similarity to AlaS. Since the GSA intermediate is unstable, GluTR and GsaM usually tend to form the stable complexes for the substrate channeling [19][6].
3. Formation of the Common Tetrapyrrole Core UPG III
UPG III, the common core of tetrapyrroles, is formed through the condensation of eight 5-ALA molecules in a highly conserved three-enzyme pathway (Figure 2). These three enzymes can be considered as the backbone of the biological tetrapyrrole synthesis tree, with a variety of branches formed by modifications and different metals [5][7].
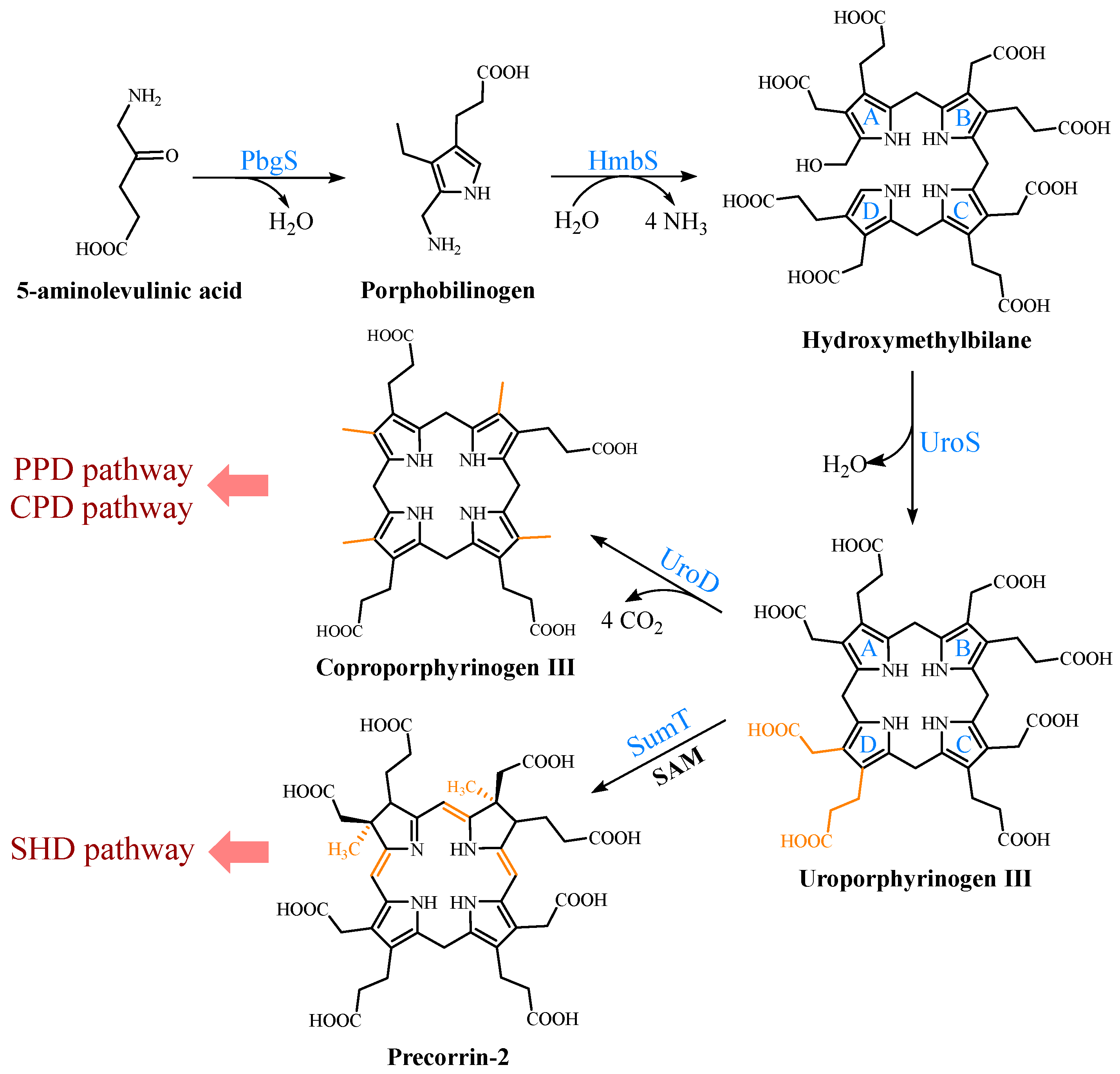
Figure 2. Formation of uroporphyrinogen III (UPG III) and the branching of the heme b biosynthesis. In nature, the pathway from 5-aminolevulinic acid (5-ALA) to UPG III is a highly conserved three-enzyme cascade. UPG III is decarboxylated by uroporphyrinogen III decarboxylase to form coproporphyrinogen III (CPG III), a reaction shared by the protoporphyrin-dependent (PPD) and coproporphyrin-dependent (CPD) pathways. In the siroheme-dependent (SHD) pathway, the C2 and C7 positions of UPG III are methylated by the SAM-dependent uroporphyrinogen III methyltransferase (SumT) to generate precorrin-2.
At the beginning, the asymmetric condensation of two 5-ALA molecules catalyzed by porphobilinogen synthase (PbgS) forms the monopyrrole porphobilinogen (PBG) [20][8]. This enzyme contains two 5-ALA binding sites, called A and P. The first 5-ALA molecule that binds to the enzyme forms the propionate side chain of PBG, while the second forms the acetate side chain [21][9]. The enzyme requires metals to be active in both the eukaryotes and prokaryotes [22][10], and the metal requirement determines the features of the A site. In the next step, the pyrrole building block PBG is used for the large-loop assembly. Hydroxymethylbilane synthase (HmbS) catalyzes the sequential deamination of the aminomethyl groups of the four PBG molecules, and the deaminated pyrroles are strung together to produce the linear tetrapyrrole hydroxymethylbilane (HMB) [23][11]. For the HmbS catalysis, two PBG molecules are required to form a unique dipyrromethane cofactor through covalent linkage, and the enzyme-bound tetrapyrrole is formed after the sequential addition of PBG, followed by the cleavage of the reaction product HMB, leaving the holo-enzyme behind [24][12]. Finally, uroporphyrinogen synthase (UroS) cyclizes HMB and notably reverses the D-ring, producing the only asymmetric isomer, UPG III [25][13]. Although a simple cyclization can produce a type I isomer, the ring reversal was favored by evolution [26][14]. UroS is highly sensitive to proteolysis and heat denaturation, resulting in a significantly lower intracellular abundance than HmbS [11][1].
4. Multiple Pathways for Synthesizing Heme b
Three heme b synthesis pathways have been discovered in living organisms (Figure 3). The PPD and CPD pathways are known as the two branches of the classical heme b synthesis pathway. The PPD pathway is found mainly in eukaryotes and many gram-negative bacteria, while the CPD pathway is found mainly in gram-positive bacteria. As shown in Figure 2, the decarboxylation of UPG III catalyzed by uroporphyrinogen III decarboxylase (UroD) [27][15] forms coproporphyrinogen III (CPG III), which is the last step before the branching pathways. In both classical pathways, the four methyl groups of heme b are produced by the stepwise decarboxylation of the acetate side chain of UPG III, with four methyl groups produced at the C2, C7, C12, and C18 positions [28][16]. Hence, no methylation of the tetrapyrrole backbone occurs in the classical pathways. The alternative siroheme-dependent (SHD) pathway is considered to be an evolutionary remnant from an anaerobic world [29][17]. Some archaea and sulfate-reducing bacteria rely on this pathway to synthesize siroheme and then convert it into heme b [30][18]. Unlike the two classical routes, the SHD pathway involves the S-adenosyl-L-methionine (SAM)-dependent methylation of the C2 and C7 positions of the tetrapyrrole backbone [31][19].
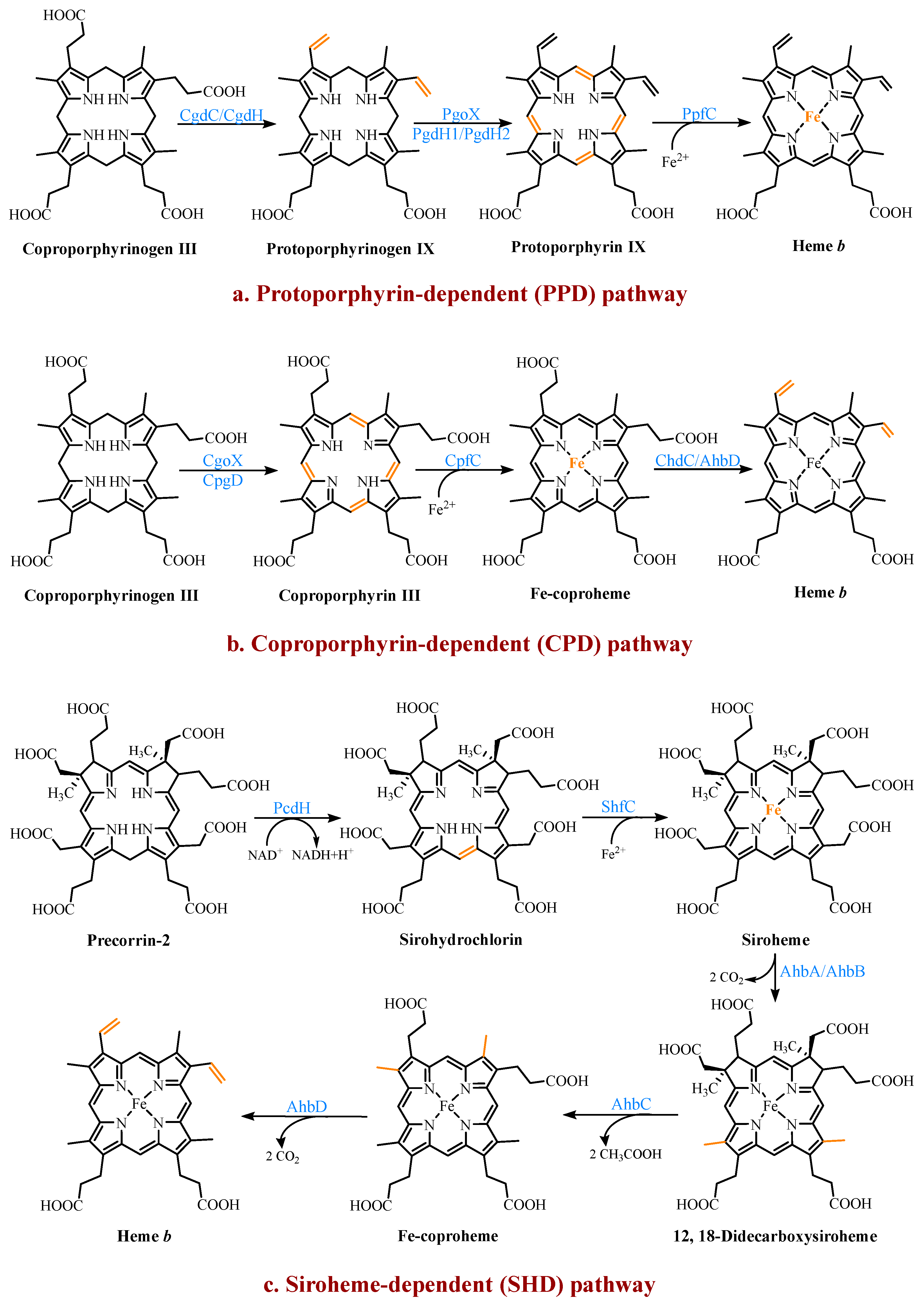
Figure 3. The three known pathways for the heme b biosynthesis. In the protoporphyrin-dependent (PPD) and coproporphyrin-dependent (CPD) pathways, the four methyl groups of heme b are the result of the stepwise decarboxylation of the acetate side chains of uroporphyrinogen III (UPG III), with four methyl groups produced at the C2, C7, C12, and C18 positions. The SHD pathway involves the S-adenosyl-L-methionine (SAM)-dependent methylation of the C2 and C7 positions of the tetrapyrrole backbone.
4.1. The Protoporphyrin-Dependent (PPD) Branch
In the PPD branch (Figure 3a), the two propionic acid side chains on the A- and B-rings of CPG III are oxidized and decarboxylated to their corresponding vinyl groups to form protoporphyrinogen IX (PPG IX) [32][20]. Two main types of enzymes catalyze this reaction. One is the oxygen-dependent coproporphyrinogen III decarboxylase (CgdC), which is present in most eukaryotes and a small percentage of gram-negative bacteria [33][21]. The other is the oxygen-independent coproporphyrinogen III dehydrogenase (CgdH), an exclusively bacterial enzyme and a member of the free radical SAM protein family [34][22]. They are completely unrelated in their structure and mechanism, but both first catalyze the modification of the A-ring and form a monovinyl monopropionate deuteroporphyrin intermediate [35][23].
Next, PPG IX undergoes a six-electron oxidation to produce protoporphyrin IX (PP IX). Three completely different enzymes have been found to catalyze this reaction, including the oxygen-dependent protoporphyrinogen IX oxidase (PgoX), and two oxygen-independent protoporphyrinogen IX dehydrogenases (PgdH1 and PgdH2). The FAD-containing PgoX shares a certain degree of sequence similarity with coproporphyrinogen oxidase (CgoX) from the CPD pathway, which is found in eukaryotes and a few gram-negative bacteria [36][24]. PgdH1 is a member of the long-chain flavodoxin family and is found mainly in gammaproteobacteria, such as E. coli. It couples the oxidation of PPG IX to the anaerobic respiratory chain rather than using oxygen directly [37][25]. PgdH2 is a membrane-bound protein, and similar to PgdH1, it does not directly interact with the electron acceptors, such as oxygen [38][26]. However, little is known about PgdH2, although it is present in almost two-thirds of gram-negative bacteria [39][27].
The final step is the insertion of a ferrous ion into PP IX, catalyzed by protoporphyrin IX ferrochelatase (PpfC). This enzyme is also poorly understood, but there is evidence that a [2Fe-2S] cluster may be a widespread feature of ferrochelatases [11,40][1][28]. However, its exact role is unclear. It is worth considering the source of the ferrous irons required by ferrochelatase due to the activity of the iron metabolism. In the organisms that were studied, there was no stoichiometric link between the iron reduction and the heme synthesis, although it was tightly linked to the cellular respiratory chain [41][29]. The complex and long pathways, such as the heme b synthesis, seem to employ multi-enzyme complexes in most organisms. Studies on eukaryotes confirmed the existence of multi-enzyme complexes containing the heme b synthetases [42][30]. However, there is still a lack of sufficient data to prove the existence of the same complexes in bacteria, and the only available data are those involving the PgoX and PpfC complexes [43][31].
4.2. The Coproporphyrin-Dependent (CPD) Branch
In evolutionary terms, the CPD pathway can be considered a transitional form between the SHD and PPD pathways [44][32]. It shares the same precursor, CPG III, with the PPD pathway, and the methyl group of the tetrapyrrole backbone is not involved in the reaction. Similar to the SHD pathway, it does not have protoporphyrinogen or protoporphyrin as intermediates (Figure 3b).
In the CPD pathway, the six electrons of CPG III are first oxidized to form coproporphyrin III (CP III). This implies that the macrocyclic oxidation in the CPD pathway occurs earlier than in the PPD pathway [11][1]. This conversion from a flexible, cyclic tetrapyrrole porphyrinogen to a fully conjugated, planar macrocyclic porphyrin is achieved through porphyrin oxidation using coproporphyrinogen oxidase (CgoX). CgoX is a soluble monomeric protein containing flavin adenine dinucleotide (FAD), which has two distinct structural differences compared to PgoX. In CgoX, the putative active-site pocket is larger and has more positively charged residues [36][24]. Based on these two features, CgoX has a higher affinity for CPG III.
The insertion of the ferrous ions to form Fe-coproheme is the next step in the CPD pathway. The iron chelatase involved in this step is coproporphyrin ferrochelatase (CpfC), which is a soluble monomeric protein [45][33]. It has an obvious structural homology with PpfC, but it can use CP III as a substrate, while PpfC cannot. This may be due to a lid consisting of a dozen residues on one side of the active site of PpfC, which can close the binding pocket during the catalytic cycle [46,47][34][35]. In this closed position, there is not enough space in the pocket to hold CP III. CpfC does not have a lid and can, therefore, accept CP III as a substrate, but this also means that its active site remains open during catalysis.
The last reaction, catalyzed using Fe-coproheme decarboxylase (ChdC), is the oxidative decarboxylation of the two propionic acid side chains on the coproheme pyrrole rings A and B to produce the corresponding vinyl groups [48][36]. The decarboxylation reaction catalyzed using ChdC requires the presence of a proton acceptor.
Under aerobic conditions, it can utilize the H2O2 produced by the earlier pathway enzyme CgoX [38][26], which is not feasible under anaerobic conditions. A gene similar to chdH, ahbD, was identified in the genomes of many gram-positive bacteria, likely encoding a free radical SAM enzyme that perhaps also catalyzes this reaction [49][37]. This enzyme will be described in detail in the section on the final step of the SHD pathway.
4.3. The Siroheme-Dependent (SHD) Pathway
The SHD pathway was only fully deciphered in 2011, confirming that siroheme is the key intermediate (Figure 3c) [30][18]. Siroheme is generated by a pathway consisting of three enzymes, including an SAM-dependent uroporphyrinogen III methyltransferase (SumT), an NAD+-dependent precorrin-2 dehydrogenase (PcdH), and a sirohydrochlorin ferrochelatase (ShfC) [50][38]. In this process, the C2 and C7 positions of UPG III are methylated to produce precorrin-2, which is then dehydrogenated to produce sirohydrochlorin, which is finally converted into siroheme through the iron insertion. In some organisms, such as E. coli, a multifunctional protein, CysG, exists, which contains the enzymatic activities of the above three enzymes and can directly convert UPG III into siroheme [51][39].
The latter three steps of the pathway are encoded using a series of ahb genes. Firstly, in the presence of AhbA and AhbB, the acetic acid side chains attached to C12 and C18 are decarboxylated to generate 12,18-didecarboxysiroheme (DDSH). The siroheme decarboxylase is either a heterodimer of the two subunits AhbA and AhbB, or a genetically encoded fusion of AhbA and AhbB. AhbC oxidizes and removes the C2 and C7 acetic acid side chains of DDSH to produce Fe-coproheme. Finally, AhbD decarboxylates the propionic acid side chains attached to C3 and C8 to produce heme b [30][18]. AhbC and AhbD both belong to the radical SAM enzyme family and, therefore, both contain an [4Fe-4S] cluster [52][40].
References
- Zhao, X.R.; Choi, K.R.; Lee, S.Y. Metabolic engineering of Escherichia coli for secretory production of free haem. Nat. Catal. 2018, 1, 720–728.
- Beale, S.I.; Castelfranco, P.A. 14C incorporation from exogenous compounds into δ-aminolevulinic acid by greening cucumber cotyledons. BBRC 1973, 52, 143–149.
- Kaufholz, A.L.; Hunter, G.A.; Ferreira, G.C.; Lendrihas, T.; Hering, V.; Layer, G.; Jahn, M.; Jahn, D. Aminolaevulinic acid synthase of Rhodobacter capsulatus: High-resolution kinetic investigation of the structural basis for substrate binding and catalysis. Biochem. J. 2013, 451, 205–216.
- Luer, C.; Schauer, S.; Virus, S.; Schubert, W.D.; Heinz, D.W.; Moser, J.; Jahn, D. Glutamate recognition and hydride transfer by Escherichia coli glutamyl-tRNA reductase. FEBS J. 2007, 274, 4609–4614.
- Ilag, L.L.; Jahn, D. Activity and spectroscopic properties of the Escherichia coli glutamate 1-semialdehyde aminotransferase and the putative active site mutant K265R. Biochemistry 1992, 31, 7143–7151.
- Nogaj, L.A.; Beale, S.I. Physical and kinetic interactions between glutamyl-tRNA reductase and glutamate-1-semialdehyde aminotransferase of Chlamydomonas reinhardtii. J. Biol. Chem. 2005, 280, 24301–24307.
- Zhang, J.; Cui, Z.; Zhu, Y.; Zhu, Z.; Qi, Q.; Wang, Q. Recent advances in microbial production of high-value compounds in the tetrapyrrole biosynthesis pathway. Biotechnol. Adv. 2022, 55, 107904.
- Breinig, S.; Kervinen, J.; Stith, L.; Wasson, A.S.; Fairman, R.; Wlodawer, A.; Zdanov, A.; Jaffe, E.K. Control of tetrapyrrole biosynthesis by alternate quaternary forms of porphobilinogen synthase. Nat. Struct. Biol. 2003, 10, 757–763.
- Coates, L.; Beaven, G.; Erskine, P.T.; Beale, S.I.; Wood, S.P.; Shoolingin-Jordan, P.M.; Cooper, J.B. Structure of Chlorobium vibrioforme 5-aminolaevulinic acid dehydratase complexed with a diacid inhibitor. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 1594–1598.
- Jaffe, E.K. The porphobilinogen synthase family of metalloenzymes. Acta Crystallogr. D Biol. Crystallogr. 2000, 56, 115–128.
- Jordan, P.M. The biosynthesis of 5-aminolaevulinic acid and its transformation into uroporphyrinogen III. New Compr. Biochem. 1991, 19, 1–66.
- Shoolingin-Jordan, P.M.; Warren, M.; Fau-Awan, S.J.; Awan, S.J. Discovery that the assembly of the dipyrromethane cofactor of porphobilinogen deaminase holoenzyme proceeds initially by the reaction of preuroporphyrinogen with the apoenzyme. Biochem. J. 1996, 316, 373–61996.
- Silva, P.J.; Ramos, M.J. Comparative density functional study of models for the reaction mechanism of uroporphyrinogen III synthase. J. Phys. Chem. B 2008, 112, 3144–3148.
- Lindsey, J.S.; Ptaszek, M.; Taniguchi, M. Simple formation of an abiotic porphyrinogen in aqueous solution. Orig. Life Evol. Biosph. 2009, 39, 495–515.
- Hashimoto, K.; Nakamura, K.; Kuroda, T.; Yabe, I.; Nakamatsu, T.; Kawasaki, H. The protein encoded by NCgl1221 in Corynebacterium glutamicum functions as a mechanosensitive channel. Biosci. Biotechnol. Biochem. 2010, 74, 2546–2549.
- Akhtar, M. Mechanism and stereochemistry of the enzymes involved in the conversion of uroporphyrinogen III into haem. New Compr. Biochem. 1991, 19, 67–99.
- Ishida, T.; Yu, L.; Akutsu, H.; Ozawa, K.; Kawanishi, S.; Seto, A.; Inubushi, T.; Sano, S. A primitive pathway of porphyrin biosynthesis and enzymology in Desulfovibrio vulgaris. Proc. Natl. Acad. Sci USA 1998, 95, 4853–4858.
- Bali, S.; Lawrence, A.D.; Lobo, S.A.; Saraiva, L.M.; Golding, B.T.; Palmer, D.J.; Howard, M.J.; Ferguson, S.J.; Warren, M.J. Molecular hijacking of siroheme for the synthesis of heme and d1 heme. Proc. Natl. Acad. Sci. USA 2011, 108, 18260–18265.
- Vévodová, J.; Graham, R.M.; Raux, E.; Schubert, H.L.; Roper, D.I.; Brindley, A.A.; Ian Scott, A.; Roessner, C.A.; Stamford, N.P.; Elizabeth Stroupe, M.; et al. Structure/function studies on a S-adenosyl-L-methionine-dependent uroporphyrinogen III methyltransferase (SUMT), a key regulatory enzyme of tetrapyrrole biosynthesis. J. Mol. Biol. 2004, 344, 419–433.
- Batlle, A.M.; Benson, A.; Rimington, C. Purification and properties of coproporphyrinogenase. Biochem. J. 1965, 97, 731–740.
- Cavallaro, G.; Decaria, L.; Rosato, A. Genome-based analysis of heme biosynthesis and uptake in prokaryotic systems. J. Proteome Res. 2008, 7, 4946–4954.
- Layer, G.; Reichelt, J.; Jahn, D.; Heinz, D.W. Structure and function of enzymes in heme biosynthesis. Protein Sci. 2010, 19, 1137–1161.
- Rand, K.; Noll, C.; Schiebel, H.M.; Kemken, D.; Dülcks, T.; Kalesse, M.; Heinz, D.W.; Layer, G. The oxygen-independent coproporphyrinogen III oxidase HemN utilizes harderoporphyrinogen as a reaction intermediate during conversion of coproporphyrinogen III to protoporphyrinogen IX. Biol. Chem. 2010, 391, 55–63.
- Qin, X.; Sun, L.; Wen, X.; Yang, X.; Tan, Y.; Jin, H.; Cao, Q.; Zhou, W.; Xi, Z.; Shen, Y. Structural insight into unique properties of protoporphyrinogen oxidase from Bacillus subtilis. J. Struct. Biol. 2010, 170, 76–82.
- Möbius, K.; Arias-Cartin, R.; Breckau, D.; Hännig, A.L.; Riedmann, K.; Biedendieck, R.; Schröder, S.; Becher, D.; Magalon, A.; Moser, J.; et al. Heme biosynthesis is coupled to electron transport chains for energy generation. Proc. Natl. Acad. Sci. USA 2010, 107, 10436–10441.
- Kobayashi, K.; Masuda, T.; Tajima, N.; Wada, H.; Sato, N. Molecular phylogeny and intricate evolutionary history of the three isofunctional enzymes involved in the oxidation of protoporphyrinogen IX. Genome Biol. Evol. 2014, 6, 2141–2155.
- Kato, K.; Tanaka, R.; Sano, S.; Tanaka, A.; Hosaka, H. Identification of a gene essential for protoporphyrinogen IX oxidase activity in the cyanobacterium Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. USA 2010, 107, 16649–16654.
- Shepherd, M.; Dailey, T.A.; Dailey, H.A. A new class of -cluster-containing protoporphyrin (IX) ferrochelatases. Biochem. J. 2006, 397, 47–52.
- Moody, M.D.; Dailey, H.A. Ferric iron reductase of Rhodopseudomonas sphaeroides. J. Bacteriol. 1985, 163, 1120–1125.
- Medlock, A.E.; Shiferaw, M.T.; Marcero, J.R.; Vashisht, A.A.; Wohlschlegel, J.A.; Phillips, J.D.; Dailey, H.A. Identification of the mitochondrial heme metabolism complex. PLoS ONE 2015, 10, e0135896.
- Masoumi, A.; Heinemann, I.U.; Rohde, M.; Koch, M.; Jahn, M.; Jahn, D. Complex formation between protoporphyrinogen IX oxidase and ferrochelatase during haem biosynthesis in Thermosynechococcus elongatus. Microbiology 2008, 154, 3707–3714.
- Dailey, H.A.; Gerdes, S.; Dailey, T.A.; Burch, J.S.; Phillips, J.D. Noncanonical coproporphyrin-dependent bacterial heme biosynthesis pathway that does not use protoporphyrin. Proc. Natl. Acad. Sci. USA 2015, 112, 2210–2215.
- Al-Karadaghi, S.; Hansson, M.; Nikonov, S.; Jonsson, B.; Hederstedt, L. Crystal structure of ferrochelatase: The terminal enzyme in heme biosynthesis. Structure 1997, 5, 1501–1510.
- Medlock, A.; Swartz, L.; Dailey, T.A.; Dailey, H.A.; Lanzilotta, W.N. Substrate interactions with human ferrochelatase. Proc. Natl. Acad. Sci. USA 2007, 104, 1789–1793.
- Medlock, A.E.; Carter, M.; Dailey, T.A.; Dailey, H.A.; Lanzilotta, W.N. Product release rather than chelation determines metal specificity for ferrochelatase. J. Mol. Biol. 2009, 393, 308–319.
- Corrigall, A.V.; Siziba, K.B.; Maneli, M.H.; Shephard, E.G.; Ziman, M.; Dailey, T.A.; Kirsch, R.E.; Meissner, P.N. Purification of and kinetic studies on a cloned protoporphyrinogen oxidase from the aerobic bacterium Bacillus subtilis. Arch. Biochem. Biophys. 1998, 358, 251–256.
- Zappa, S.; Li, K.; Bauer, C.E. The tetrapyrrole biosynthetic pathway and its regulation in Rhodobacter capsulatus. Adv. Exp. Med. Biol. 2010, 675, 229–250.
- Bali, S.; Palmer, D.J.; Schroeder, S.; Ferguson, S.J.; Warren, M.J. Recent advances in the biosynthesis of modified tetrapyrroles: The discovery of an alternative pathway for the formation of heme and heme d 1. Cell Mol. Life Sci. 2014, 71, 2837–2863.
- Stroupe, M.E.; Leech, H.K.; Daniels, D.S.; Warren, M.J.; Getzoff, E.D. CysG structure reveals tetrapyrrole-binding features and novel regulation of siroheme biosynthesis. Nat. Struct. Biol. 2003, 10, 1064–1073.
- Smith, M.A.; King, P.J.; Grimm, B. Transient-State kinetic analysis of synechococcus glutamate 1-semialdehyde aminotransferase. Biochemistry 1998, 37, 319–329.
More