Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Fernando Baquero and Version 3 by Rita Xu.
The concept of “structural epistasis” expresses the emergence of new phenotypes which are not based on changes in the products and functions of genes, but on the changes in the physical–mechanical interactions between biological structural pieces and components of the bacterial cell architecture.
- bacterial Gram-negative subcellular architecture
- structural epistasis
- cellular shape and volume
1. Introduction
Etymologically, the term “epistasis” means the “act of stopping” (any “on–off” action) and can be applied to the case where a mutation influences the effect of other mutations, in a specific or unspecific and less evolutionarily stable manner [1][2][1,2]. Epistasis is also a phenomenon in which one or more genes influence the function of others. The related term “epigenetics” refers to studies “above the gene” and the heritable (reproducible) changes in gene function that cannot be explained by DNA sequence mutations [3]. In both cases, the organism exhibits a function that cannot be fully explained by the sequence of a single gene. The most classic cases of epistasis rely on the interconnection of regulatory networks. For instance, a mutation that alters the concentration of a metabolite that regulates the expression of other metabolic routes may deeply alter the bacterial physiology, thereby modifying the activities of other genes/proteins. Examples of this include the modification of the physiology of antibiotic-resistant mutants [4][5][4,5] or the various evolutionary pathways toward resistance that bacteria with differing genomic backgrounds can follow, which is an example of historical contingency [6]. However, these regulatory and functional alterations can have an important structural role, a feature largely underexplored.
In general, and in the abovementioned epistatic examples, a phenotype is the result of the interconnected functions of an ensemble of genes (not necessarily linked), as in the case of an operon encoding for antibiotic resistance, or in biosynthetic pathways. In the last case, however, the function of each involved gene is autonomous and specific; for instance, encoding an enzyme needed for a particular biochemical reaction. The operational molecules involved in epistasis are the products of interacting genes: proteins interact with other proteins, small molecules or nucleic acids. In Escherichia coli, the concentration of these macromolecules can exceed 300 g/L, occupying approximately 30% of the cell volume [7][8][7,8].
In population genetics, epistasis refers to how genetic interactions between some loci affect phenotypes and fitness [9]. The concept of structural epistasis helps explain the emergence of new phenotypes that are not based on changes in gene function, but on the physical–mechanical interactions between the biological “structural pieces” or components of the cell. These interactions result from primary physical changes in the piece’s shape and size, or in the spatial (topological) alterations driven by changes in their quantity or local density. This might result in spatial heterogeneities leading to various cell-to-cell physical interactions, that also have consequences in microbial cell biology. The term “cell topology” is employed in studies on the structure of tissues, where spatial heterogeneity might produce differing biological outcomes [10]. Starting about one decade ago, new technologies have become available to study intracellular topology, as multi-scale fluorescence cross-correlation spectroscopy [11].
A bacterial cell is composed of complex physical multimolecular objects, which include: (1) ball-shaped complex structures, such as ribosomes, supercoiled DNA in the chromosome (forming a nucleoid) or in bacterial plasmids; (2) lamellar structures, such as the cell wall, membranes or capsules; (3) elongated structures, such as fimbriae and flagella; (4) complex-shaped functional organelles, ranging from low complexity, such as porins, to high complexity, such as extrusion pumps, needle-like protein complexes in the type III secretion system, or trans-envelope flagella machines; and (5) ball-shaped inclusion bodies, typically water-insoluble protein aggregates or condensates, glycogen poly-beta-hydroxybutyrate granules, or cyclic polyphosphate inclusions. These complex structures in the bacterial cytoplasm, and the molecular crowded cytoplasm itself, have inherent physical properties, such as viscoelasticity [12] or electric charge, which affect their function [13]. Molecules have both a physical and chemical dimension. For instance, the interaction and function of proteins depends on their tertiary structure, the three-dimensional arrangement (folding) of its polypeptide chain in space, and on their four-dimensional (protein-assembly in multimeric proteins) and fifth-dimensional (quinary structure, see below) structures, ensuring the interactions between macromolecules that organize the interior of the cell.
Similarly and most importantly, DNA topology influences replication and gene expression. The bacterial chromosome is a highly structured molecule organized into various domains (including macrodomains and replichores) showing varying degrees of gene expression, supercoiling, protein occupancy, and the binding of xenogeneic silencing proteins (e.g., H–NS, histone-like nucleoid-structuring proteins) [14][15][16][14,15,16]. All these physical objects are arranged in a physiological intracellular “ecological topology”, understood as the pattern of interconnections of a molecular network, and based on physical (structural) properties that vary over time (according to growth phases) and should be tightly regulated [17]. For instance, the cell should manage the encounters between replication and transcription machineries, given that conflicts between them can lead to genome instability and reduced fitness [18].
In turn, healthy topology contributes to molecular (e.g., rRNA, proteins, nutrients) mobility and interaction, at times mediated by particular compounds, such as histone-like nucleoid-structuring H–NS proteins, which interact with both proteins and DNA [16]. The bacterial cell therefore exhibits a well-controlled spatial organization, with particular degrees of flexibility (adaptive organization) that are beginning to be explored [19][20][19,20]. There are precedents for such a type of exploration in the morphogenetic studies of the past century. In 1952, Alan Turing (the godfather of modern computing) proposed that biological morphogenesis could be explained by an stochastic activator–inhibitory system, that gives rise to particular organizational patterns, with modern modeling studies showing the plausibility of this approach [21]. In the following section, a succinct description of the architectural components of the bacterial cell is presented (Figure 1).
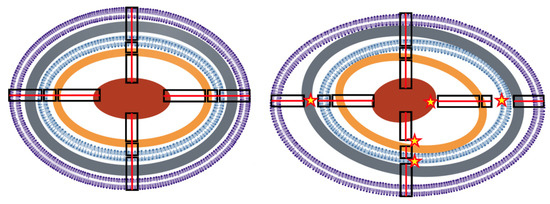
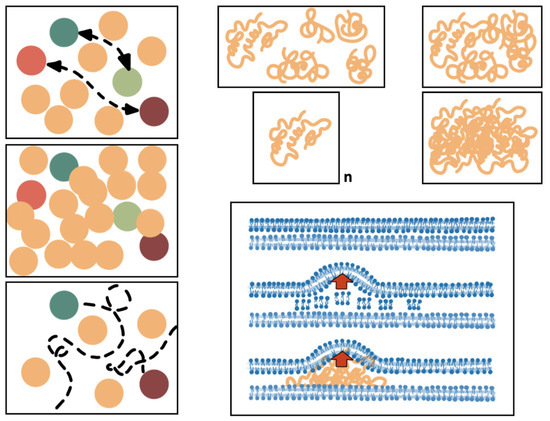
Figure 1. Conceptual illustration of structural epistasis. (Left) Simplified concentrical structure of a normal Gram-negative bacterium. From the outside to inside, in violet, the bilayer outer membrane (OM); in grey, the peptidoglycan; in blue, the bilayer internal membrane (IM); in brown, the ribosome-dense crown; in red, the nucleoid. Proteins (not represented) are particularly dense among the most external layers. Black rectangles illustrate structural connections across layers (for simplicity only four are depicted), successively, OM–IM, IM–ribosomes, ribosomes–nucleoid. Continuous red lines across the connections illustrate the integrity of the structure. Physiological changes are not expected to eliminate the concentrical structure, as during division, new centers are created. (Right) A distorted cellular structure, resulting from exposure to stressful conditions, where the layers are losing their normal connectivity (stars) producing wider spaces in some areas and narrower spaces in others. The result of the architectonical distortion influences the relative molecular interactions (interrupted red lines), changing the local protein densities and interactions (see Figure 2).
Figure 2. Effect of structural distortion in the protein–protein and protein–OM interactions. The upper left panel shows a normal density of the proteins (ovals) in a structured space; functional connections (double-headed broken arrows) between particular proteins (light–dark red and light–dark green) occur normally. If the protein density in very high (mid left panel), these interactions might be prevented. On the contrary, if the density is too low (lower left panel), the connections cannot be established. The upper right panels illustrate the 3D folding of proteins (orange crumpled lines). If the protein density is increased, the proteins might interact and eventually change their shape non-specifically. Eventually, conglomerates of the same protein might occur, producing protein inclusion bodies. The lower right panel exemplifies the distortion of the normal topology of the OM (up in the panel) due to the overproduction of membrane components (middle panel) or the location of a protein inclusion body (e.g., an hyperproduced protein, such as a beta-lactamase). In both cases, the distortion of the OM (red arrow) might alter the location/interaction of particular proteins (including porins, specific receptors, and pumps proteins, not represented for simplicity), which results in altered phenotypes.
2. The Bacterial Cell Architecture and Shape Is Altered by Antibiotic Resistance
Antibiotics frequently promote (or select for) a few phenotypic variants in the population (known as “persisters”) that tolerate antibiotic action at the expense of changing the cellular architecture, resulting in altered cell shapes. In E. coli tolerating ampicillin exposure, the persisters are frequently smaller and the cells are swollen, altering physical molecular interactions by changing elasticity or surface-to-volume ratio [22][72]. In Staphylococcus aureus, antibiotic persisters are usually recognized as “small colony variants”, frequently changing the envelope (thicker cell walls) and, correspondingly, a denser granulation in the peripheral cytoplasm, or have branched or multiple cross walls, which are sometimes defective [23][73]. In addition, exposure to bacteriostatic agents, such as macrolides and chloramphenicol, results in small colony variants [24][74]. Given that it also occurs in pH-driven transitions, bacterial dormancy or persistence could possibly be due to a transition of the cytoplasm from a fluid to a gel-solid-like state [25][26][75,76].
Changes in cell shape induced by antibiotic action have real consequences for the molecular mobility inside the cells (and consequently metabolic activity), which has been tested by using the intracellular diffusion of green fluorescent protein (GFP) in the presence of antibiotics [27][28][29][60,77,78]. The hyperexpression of resistance mechanisms might also affect bacterial architecture. In general, protein hyperexpression leads to the formation of small colony variants and reduces bacterial growth, as was signaled in recombinant strains modified to hyperproduce (for instance using strong promoters) proteins for chemical or pharma industries. To address this problem, the shape of the bacteria should be modified (through morphology engineering) [30][79]. High levels of TEM-1 beta-lactamase expression results in sequestration of the mature excreted polypeptide in insoluble protein aggregates (inclusion bodies) located within the periplasmic space. Given that correct protein folding is altered in these aggregates, the function is lost [31][80]. The constitutive hyperexpression of the AmpC-type beta-lactamase might also have a deleterious effect on cellular fitness [32][33][81,82]. Studies on Salmonella, the only Enterobacteriaceae that does not possess AmpC, have shown that the acquisition of exogenous AmpC causes changes in cellular morphology, with longer cells indicating an effect on septation, and produces small changes in the structure of the peptidoglycan. These effects fully abolish Salmonella Typhimurium viability, unless ampC expression is under control of its regulator, AmpR [32][81]. A similar effect of a severe fitness cost occurs in Pseudomonas aeruginosa [33][82]. The metallo-carbapenemase NDM-1 is related with lipoproteins of the OM and can be packaged into the periplasmic space by physical interactions with the OM layer, determining “envelope stress” and facilitating its extrusion inside membrane vesicles [34][83]. MDR efflux pump hyperexpression should also physically disturb the anatomical and functional structure of the cell envelope, with consequent effects of an altered cell physiology. However, this structural disturbance is still an underexplored area. The fact that particular Stenotrophomonas maltophilia resistant mutants present a reduced cell size and a lower plate efficiency might shed light the direction of future studies [35][84].
Nevertheless, efflux pump overexpression modifies the activity of other unrelated cell machineries, thereby providing an example of structural epistasis. This is the case for MexEF-OprN in the Pseudomonas aeruginosa, of which the overexpression increases oxygen respiration, modifies the intracellular pH and triggers the activation of the nitrate respiratory pathway [36][85]. Notably MexEF-OprN overexpression is associated with a reduction in type III secretion [37][86], and type III secretion requires significant activity of the proton motive force [38][87]. Whether MexEF-OprN overexpression inactivates type III secretion by modifying the proton motive force, thereby impeding the simultaneous induction of these two energetically costly cell machineries, and if the reduction in fitness is also influenced by less efficient basic metabolic pathways (as energy in invested in overexpression of the secreted protein), are questions that remain unanswered.
The conjugative process of plasmids (which eventually encode antibiotic resistance) might produce (probably small) alterations in the bacterial architecture of the donor cell, because of the relaxosome protein complex (docked to the Type IV secretion system) and the entire conjugative apparatus, forming an envelope structure bridging the IM and the OM [39][88]. In the donor and recipient cells, the exit and the entry of the ssDNA plasmid triggers the local recruitment of Ssb (single-strand binding protein) molecules and the formation of membrane conjugative foci, which are apparently located at specific membrane positions, possibly related to the density and stability of the outer membrane protein OmpA, a beta-barrel porin collaborating in the process [40][89]. Eventually, mobile genetic elements (preferentially small plasmids) might contribute to increasing particular gene dosages, and thus gene dosage toxicity is based on abnormal protein abundance levels. Dihydrofolate reductase overexpression in E. coli causes a metabolic imbalance, reducing bacterial fitness [41][90].
3. Cell Architecture and Cell Size Influences Antibiotic Effects
Any change in the cell shape requires an expansion or constriction of the membrane layers (and the periplasm volume), which involves a quantitative change in their molecular components or in the distance between them, and therefore their physical interaction. The concept of structural epistasis is applied here to discuss whether these changes might have functional consequences, including the susceptibility to antimicrobial action [42][66]. Quantitative modeling has shown that bacteria might adapt (reduce their susceptibility) to antibiotic challenges by reducing the surface-to-volume (S/V) ratio, and consequently, the antibiotic influx and intracellular concentration. In certain cases, however, increasing this ratio might provide an increase in antibiotic efflux rate, and thereby reduced susceptibility. Moreover, the concentration of membrane-associated antibiotics is reduced [43][65]. In fact, most tested antibiotics decrease the S/V ratio. Such a reduction also decreases nutrient uptake, slowing the bacterial metabolism and consequently the antibiotic action [44][91]. As expected, the morphological cell response to membrane-targeting and membrane-transport-targeting antibiotics is a reduction in the cell surface area. Other effects of the S/V can be considered secondary to the bacterial adaptation to drug action; for instance, the inhibition of translation by ribosome-targeting antibiotics (as chloramphenicol) is compensated by a higher ribosomal biosynthesis, leading to the predominance of growth versus replication, with the consequences of an increased cell volume [44][91].
Membrane changes are not necessarily global. Different bacterial processes are confined to membrane microdomains that are similar to lipid eukaryotic cell lipid rafts, which accumulate multimeric protein complexes favoring their oligomerization. One of these proteins, PBP2a, causes beta-lactam resistance in methicillin-resistant Staphylococcus aureus (MRSA). Notably, the disruption of these membrane microdomains with available drugs (such as statins, regularly used for treating hypercholesterolemia) interferes with PBP2a oligomerization, resulting in MRSA infections that are treatable with penicillin [45][92]. This example shows that structural alterations in a cellular element (the cell membrane) impedes the structural changes (oligomerization) required for the activity of a wide-spread antibiotic resistance gene.
In addition, bacteria readily alter their shape in response to non-antibiotic cues. For instance, during urinary tract infections, E. coli produces long multi-nucleated filaments in response to a still unknown urine component [46][93]. In addition to decreasing the S/V ratio, filamentation has numerous consequences for the bacterial lifestyle. For instance, filamented bacteria are less likely to be attacked by phagocytes [47][94], and thanks to the extra body-mass, filamented bacteria might have an improved ability to resist shear forces in the bladder and adhere to the epithelium [48][49][95,96]. Beyond the urinary tract, filamentation allows intracellular bacteria to spread among host cells [50][97] and might promote the evolution of antibiotic resistance [51][98], which provides an excellent example of how physical changes in cell structure can shape numerous, non-related phenotypes.