Calcium (Ca2+) is a divalent cation and a universal second messenger that regulates the most important functions and facets of all eukaryotic cells, including: gene expression, proliferation, regulation of bioenergetics, contraction of muscles, mediation of fertilization, and many other cellular functions. Regulation of free intracellular concentration of Ca2+ is an important mechanism for intracellular signaling, and it is a key component in the mediation of many cell functions and biochemical reactions, being crucial for signal transduction in cells. On top of all that, intra-mitochondrial Ca2+ regulates a cascade of physiological and pathophysiological processes in cells The normal level of intra-mitochondrial Са2+ is essential for the correct functioning of mitochondria; whereas Ca2+ overload is typical for a wide range of mitochondrial dysfunctions and pathophysiological processes. Homeostasis of Ca2+ in the mitochondria is determined by the delicate balance of mitochondrial Ca2+ transport systems in both the inner (IMM) and outer mitochondrial membrane (OMM). Ca2+ influx and efflux systems are composed of different components, including: channels, pumps, antiporters, or Ca2+ binding proteins that cooperate to maintain intra-mitochondrial Ca2+ homeostasis.
- mitochondria
- Calcium transport
- VDAC
- MCU
- RaM
- mRyR
- mPTP
- LETM1
- NCLX
- HCX
1. Introduction
The organelles responsible for Ca2+ homeostasis are undoubtedly the mitochondria, which are essential for cellular bioenergetics, not only by storing energy in the form of ATP, but also by playing a major role in Ca2+ signaling [15][212][316][417]. Ca2+ uptake by mitochondria not only participates in the regulation of cytosolic Ca2+ concentration ([Ca2+]), but also stimulates mitochondrial respiration and ATP production[518][619]. These properties make these organelles the major cellular components in the regulation of the fate of a cell[79][212][417][820][921].
Localization of mitochondria inside the cell can vary significantly: from the periphery of the cell, around the nucleus, but also close to the plasma membrane or the endoplasmic/sarcoplasmic reticulum (ER/SR)[1022][1123]. These different localizations determine the Ca2+-buffering capacity of each individual mitochondria as well as the mitochondrial network[212]. Upon contact of the mitochondria, or more specifically the outer mitochondrial membrane (OMM) with other organelles, membrane contact sites are formed[1224]. These inter-organelle associations have various functions. For instance, those formed between the mitochondria and ER/SR (mitochondria-associated membranes, MAMs) determine Ca2+-uptake from the cytoplasm to the mitochondria, and therefore play an essential role in the Ca2+ signaling pathways[1123][1325][1426]. It is established that such associations contain microdomains with high Ca2+ concentrations that determine the mediation of Ca2+ transport between the mitochondria and the ER/SR [24]. Moreover, mitochondrial associations with the plasma membrane are engaged in the mediation of Ca2+ transport from the extracellular environment[1527].
Mitochondria are the power generators of cells. They produce ATP in the citric acid cycle (the tricarboxylic acid (TCA) or the Krebs cycle (see Box 1 for more information). Production of ATP involves activation of the Ca2+-dependent dehydrogenases in the citric acid cycle, F0F1-ATP-synthase and metabolite transporters; all of them being supplied by basal oscillating increases in the concentration of Ca2+ in the mitochondrial matrix[166][1728][1829][1930]. In addition to these normal physiological oscillations, large Ca2+ spikes in mitochondria can cause an opening of the mitochondrial permeability transition pore (mPTP)[79][1728][1829]. In turn, this induces a collapse of the mitochondrial membrane potential, termination of oxidative phosphorylation processes, osmotic changes, mitochondrial swelling, and inner membrane remodeling. All of these processes culminate by mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome c; being both an inducer of apoptosis and modulator of other proapoptotic factors[202][212][2131][2232]. Whereas mostly associated with programmed cell death, a number of compounds trigger changes in Ca2+ homeostasis and mPTP-induced apoptosis[202][79][1829].
The ability of mitochondria to uptake and retain Ca2+ had already been described in the early 1960’s using isolated mitochondria[2333][2434]. During the same years, the chemiosmotic theory, as proposed by Mitchell[2535], revealed the thermodynamic basis of the process.
|
Box 1. The generation of ATP by mitochondria Mitochondria are the power generators within all eukaryotic cells. They release their energy in the form of ATP by the oxidation of sugars. Electrons supplied by NADH are transferred to oxygen by a series of protein complexes in the inner mitochondrial membrane. By pumping protons across the membrane, these complexes create a transmembrane electrochemical gradient (ΔΨ, ~ -180 mV). This reverse current of protons into the mitochondrial matrix occurs through a proton channel formed by ATP synthase, and it is used to store energy in the form of ATP. |
Mitochondria are able to rapidly accumulate and transiently store Ca2+ for later quick release, making these organelles important cytosolic depots or buffers for Ca2+ regarding mediation of the cell's physiological and pathological processes, including from cell survival to cell death[263][277][212][921][1426][2837]. Regulated elevations of Ca2+ levels in the mitochondrial matrix are necessary for the regulation of Ca2+-dependent mitochondrial enzyme activity, which sequentially mediates the metabolic balance and function of the mitochondrial electron transport chain, as well as the production of mitochondria-generated reactive oxygen species (ROS)[212][2938][3039][3140]. Undoubtedly, the precise regulation of mitochondrial Ca2+ uptake and release is necessary for proper cellular functioning and regulation of mitochondrial bioenergetics. The normal level of intra-mitochondrial Са2+ is essential for the correct functioning of mitochondria; whereas Ca2+ overload is typical for a wide range of mitochondrial dysfunctions and pathophysiological processes[3214][3315][2837][3039][3441]. Homeostasis of Ca2+ in the mitochondria is determined by the delicate balance of mitochondrial Ca2+ transport systems in both the inner (IMM) and outer mitochondrial membrane (OMM) (Figure 1). Ca2+ influx and efflux systems are composed of different components, including: channels, pumps, antiporters, or Ca2+ binding proteins that cooperate to maintain intra-mitochondrial Ca2+ homeostasis[3510][3214][2938][3039][3642].
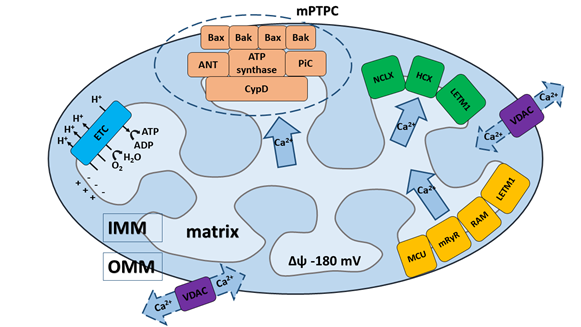
Figure 1. Schematic presentation of Ca2+ transport systems in mitochondria. (1) Ca2+ influx and efflux through the outer mitochondrial membrane (OMM) driven via the voltage-dependent anion channel (VDAC). (2) Ca2+ influx through the inner mitochondrial membrane (IMM) driven by three major transport systems: (i) mitochondrial Ca2+ uniporter (MCU), (ii) mitochondrial ryanodine receptor (mRyR), (iii) rapid mode of Ca2+ uptake (RaM) and one mitochondrial system under debate: (iv) leucine zipper- EF-hand containing transmembrane protein (LETM1). Ca2+ influx through the MCU is established by the electrochemical gradient created by the electron transport chain (ETC). (3) Ca2+ efflux through the IMM driven by three other major transport systems: (i) Na+/Ca2+/Li+ (NCLX) exchanger, (ii) H+/Ca2+ exchanger (HCX), (iii) mitochondrial permeability transition pore complex (mPTPC) and one mitochondrial system under debate: (iv) leucine zipper- EF-hand containing transmembrane protein (LETM1). (4) The core constituents of mPTPC include: the adenine nucleotide translocase (ANT), matrix cyclophilin D (CypD) and phosphate carrier (PiC), which serve as pore regulators, and the pro-apoptotic proteins Bax and Bak, which can induce mitochondrial swelling and rupture during the mPTP opening. ATP-synthase is the key IMM-pore forming unit of mPTPC.
2.1 Calcium influx and efflux through OMM
When Ca2+ enters the mitochondrial matrix from the cytoplasm, it first encounters the OMM. This membrane is highly permeable to cations, anions, and molecules with molecular weights <5 kDa due to the presence of large conductance channels. These channels, formed by voltage-dependent anion channel proteins (VDACs), allow for the exchange of Ca2+ and small molecules by concentration gradients[202][3743][3844][3945][4046][4147]. They not only regulate transport of Ca2+ from the cytoplasm into the intermembrane space (IMS), but are additionally engaged in the mediation of cellular metabolism by transporting ATP and other small metabolites across the OMM[212][4248]. Importantly, the permeability of VDACs is precisely controlled and regulated, particularly by ATP and a variety of cellular regulatory factors.
VDAC (Figure 1) was the first channel that has been reconstituted and characterized in detail at the single-channel level[3844][4349]. It has been proposed to work as the principal metabolite transport system across the OMM, and had also been proposed to serve as the interconnection point between the OMM and IMM[4450]. Later, three different VDAC isoforms were identified: VDAC1, VDAC2, and VDAC3[202][3844][4147][4551].
VDAC1 is highly expressed in most cells, and seems to be the most prevalent and most extensively characterized; it is also considered as the main transport channel for Ca2+[202] [4652]. VDAC1 is the gatekeeper for the passage of ions and metabolites, and is crucial for the regulation of apoptosis, thanks to its interactions with pro- and anti-apoptotic proteins[1123][4753]. Activity of VDAC1 is critical for the mitochondrial metabolic pathways balance, as well as for cell survival[4753]. Imaging of VDAC1 by stimulated emission depletion nanoscopy revealed the organization of VDAC proteins into clusters in H9C2 cells, which has also been studied in VDAC transfected U2OS cells[4854][4955]. VDAC1 consists of 19 transmembrane β-strands that are organized into the membrane-incorporated β-barrel and a amphipathic 26-residue-long N-terminal domain, which can translocate from the pore interior to the channel surface[5056]. This behavior is crucial for controlling the gating of the channel as well as its interactions with apoptotic proteins[5056][5157]. Whereas isoforms of VDAC1 and VDAC2 self-assemble into structures resembling a pore, VDAC3 forms smaller conductance channels that are able to modulate the physiological functions of various proteins[5258]. As demonstrated by Checchetto et al. [58], VDAC3 isoforms demonstrate different electrophysiological properties compared with those of VDAC1 and VDAC2. In the context of their structural/functional characteristics, VDAC1, VDAC2, and VDAC3 have some similarities; at the same time, they exhibit different physiological functions regarding their interaction with cytosolic proteins and other mitochondrial proteins[202][4147][5359]. Furthermore, only limited information is available regarding the potential functions of VDAC2 and VDAC3 for the influx of Ca2+[3743] [5359][5460].
2.2 Calcium influx through IMM
Compared to the OMM, the IMM exhibits a fundamentally higher selectivity for anions and cations thanks to the presence of highly-specific and different protein machinery in the IMM. The key transporters that determine Ca2+ uptake by mitochondria through the IMM until recently were unclear. It is now believed that the transport of Ca2+ through the IMM is accomplished by a group of mitochondrial Ca2+ uptake transporters. Basically, three main mechanisms of Ca2+ influx have been proposed (Figure 1): (1) a mechanism that requires an electrogenic mitochondrial Ca2+ uniporter multi-protein complex (MCU complex); (2) a so-called rapid mode (RaM); (3) a mechanism requiring the mitochondrial ryanodine receptor (mRyR)[277][3510][212][5513][2938]; and (4) additionally, leucine zipper-EF-hand containing transmembrane protein (LETM1) could represent another Ca2+ influx mechanism, but its role is still under discussion[5661][5762][5863][5964].
2.2.1 Calcium influx by mitochondrial Ca2+ uniporter (MCU) multi-protein complex
The molecular identity of this Ca2+ transport pathway had been unclear for several decades. However, in 2011, the CCDC109a gene, a pore-forming component of the MCU channel, mediating Ca2+ influx into mitochondria was discovered [65,66]. The protein encoded by the CCDC109a gene is responsible for Ruthenium Red-sensitive mitochondrial Ca2+ uptake. Currently, accumulation of Ca2+ through the MCU multi-protein complex is the most widely characterized and commonly accepted pathway of Ca2+ influx into mitochondria; and it is considered as the major pathway of the mitochondrial Ca2+ influx. It is determined by a large electrochemical gradient (∼−180 mV) across the IMM, and may be inhibited by Ruthenium Red and lanthanides[277][5513][3642][6067][6168][6269]. The complex consists of several subunits, including transmembrane core components and regulatory subunits that are associated with the membrane. The core components of the MCU multi-protein complex (see Box 2 for details) are comprised of: a) core protein components: Mitochondrial Ca2+ Uniporter (MCU), a MCU dominant negative beta subunit (MCUb), and Essential MCU REgulator (EMRE); and b) membrane associated regulatory components: mitochondrial Ca2+ uptake protein 1-3 (MICU1-3) and Mitochondrial Ca2+ Uniporter Regulator 1 (MCUR1)[212][5513][1123][3642][4147]][6168][6269][6370][6471][6572]. Solute Carrier 25A23 (SLC25A23)) was initially identified as an essential component of MCU, however, it is currently under debate whether SLC25A23 is an component of MCU or whether it influences MCU indirectly [5513][3642][6673]. Importantly, the MCU complex can be found in multiple states. [60][61][62][63][64][65][66][67][68][69][70][73][76][79][80][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98][99]
|
Box 2. Structure of the MCU multi-protein complex Core components MCU (mitochondrial Ca2+ uniporter, previously known as CCDC109a, 40kDa) is a key core component of the complex. It is encoded by a highly conservative MCU gene and is present in virtually all eukaryotic organisms [3510][5513][4147]. MCU can be found in multiple states, and it consists of two coiled-coil domains (CC) and two transmembrane domains connected via a short loop (9 amino acid residues) containing a highly conserved DIME motif [42,65,66]. MCUb (MCU dominant negative beta subunit, formerly known as CCDC109b, 40 kDa) is a core component of the MCU multi-protein complex encoded by the MCUb gene, and is present in all vertebrates[6471][6572][6774]. It exhibits a 50% homology with MCU; however, MCU and MCUb demonstrate diverse expression profiles in different tissues. Importantly, MCUb significantly impairs Ca2+ permeation through MCU[3642][6269][6370]. EMRE (essential MCU regulatory element, 10-12 kDa) is the last core component identified in the complex. It contains a single transmembrane segment, and crucially regulates MCU activity as has been shown using EMRE knockout cells, which inhibited mitochondrial Ca2+ influx[6875]. EMRE is assumed to be involved in the formation of interactions between the core and the regulatory subunits, despite the fact that such ensembles of regulatory components do not require the presence of EMRE[5513][3642][4147] [6875]. Membrane-associated regulatory components MICU1 (mitochondrial Ca2+ uptake protein 1, known as CBARA1/EFHA3, 54kDa) known as CBARA1/EFHA3, is a membrane-associated and water-soluble component localized in the inter-membrane space; it is considered as central for the activation of MCU. In the resting state (i.e., at low intracellular concentrations of Ca2+), MICU1 blocks access of Ca2+ to the MCU channel[6875][6976][7077][7178]. It also acts as a cooperative activator of MCU and it stimulates MCU Ca2+-transport conductivity[6976]. MICU2 (mitochondrial Ca2+ uptake protein 2, known as EFHA1, 50 kDa) and MICU3 (mitochondrial Ca2+ uptake protein 3, known as EFHA2, 60 kDa) display the EF-hand domains in the protein structure, and were identified as MiCU1 paralogs with 41% and 34% identity to the MICU1, respectively[71][72] [78–80]. MiCU2 forms heterodimers with MiCU1 through disulfide bonds, and acts as a Ca2+ sensor, protecting the mitochondria against Ca2+ overload, and it also acts as the regulator of several cell functions[7376]. MiCU2 forms heterodimers with MiCU1 through disulfide bonds, and acts as a Ca2+ sensor, protecting the mitochondria against Ca[81]2+[82] overload[76,81, and it also acts as the regulator of several cell functions[69][74][75].82]. MCUR1 (mitochondrial Ca2+ uniporter regulator 1, known as CCDC90A, 40 kDa) is composed of 2 transmembrane domains and 1 specific coiled-coil region, and it belongs to yet another regulatory component of the MCU complex[76][77] [83,84]. MCUR1 knockdown prevents Ca2+ entry into the mitochondria; whereas, its overexpression promotes mitochondrial Ca2+uptake [82,84]. MCUR1 kinteracts with EMRE anockdown prevents Cd MCU-pore via its coiled-coil domains, which stabilize all components of the MCU complex [85]. It is involved in the assembly of the mitochondrial respiratory chain, and represents a cytochrome c oxidase assembly factor; possibly also regulating the mitochondrial membrane potentia2+ entry into the mitochondria; whereas, its overexpression promotes mitochondrial Ca2+uptake[75][77]. MCUR1 interacts with EMRE and MCU-pore via its coiled-coil domains, which stabilize all components of the MCU complex[78]. It is involved in the assembly of the mitochondrial respiratory chain, and represents a cytochrome c oxidase assembly factor; possibly also regulating the mitochondrial membrane potential[79].l [86]. SLC25A23 (solute carrier 25A23, 48-54 kDa) was initially identified in the IMM as a protein with the EF-hand domain, and has been proposed as a component of MCU multi-protein complex[20][79][80]. [2,86,87]. SLC25A23 may also function as an ATP-Mg/Pi exchanger, promoting the influx of adenine nucleotides into the matrix of mitochondria and the efflux of inorganic phosphate. Of note, SLC24A23 functions in a Ca2+ dependent manner [73,88]. Mutations and modifications of the EF-hand domains in this carrier decrease Ca2+ influx into mitochondria; however, it still remains unclear whether SLC25A23 influences the uniporter complex directly or whether it affects the mitochondrial bioenergetics [13,42,87]. Further studies are necessary to understand the exact mechanism by which SLC25A23 regulates mitochondrial Ca2+ SLC25A23 may also function as an ATP-Mg/Pi exchanger, promoting the influx of adenine nucleotides into the matrix of mitochondria and the efflux of inorganic phosphatenflux. Of note, SLC24A23 functions in a Ca2+ dependent manner [73,88]. Mutations and modifications of the EF-hand domains in this carrier decrease Ca2+ influx into mitochondria; however, it still remains unclear whether SLC25A23 influences the uniporter complex directly or whether it affects the mitochondrial bioenergetics[55][36][80]. Further studies are necessary to understand the exact mechanism by which SLC25A23 regulates mitochondrial Ca2+ influx. |
2.2.2 Rapid mode mechanism (RaM) of Ca2+ uptake
The RaM (RApid Mode of Ca2+ uptake) mechanism is able to accumulate Ca2+ up to a hundred times faster compared with the MCU multi-protein complex (no molecular structure responsible for this mechanism has yet been identified)[81][82] [89,90]. It is transiently activated by low calcium concentrations (50-100 nM) and by high concentrations of Ruthenium Red[55][82][83] [13,90,91]. This behavior contrasts sharply with MCU, which is activated by Ca2+ concentrations higher than 500 nM. RaM promotes mitochondria to rapidly sequester Ca2+ at the beginning of each cytosolic Ca2+ pulse, and rapidly recovers between pulses, allowing mitochondria to respond to repetitive Ca2+ oscillations[55][83] [13,91]. It is still speculated that RaM is just an additional state of the MCU multi-protein complex because of their similarity as well as the absence of RaM in MCU knockout mitochondria[84][82][83] [65,90,91]. At present, the progress of research targeted on explaining the role of RaM in Ca2+ influx at the molecular level is very limited.
2.2.3 The mechanism of Ca2+ uptake requiring mitochondrial ryanodine receptor (mRyR)[3][4][5][6][7][8][10][11][13][14][15][16][25][26][29][30][31][32][33][34][35][36][37][38][39][40][41][42][12][44][45][46][47][48][49][50][2]][51][52][53][54][55][56][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][57][23][1]
mRyR (mitochondrial ryanodine receptor, 600 kDa) is the ryanodine-sensitive mitochondrial Ca2+ uptake mechanism, capable of Ca2+ transport, which was detected in the IMM of isolated heart mitochondria in 2001 by Beutner at al. [92]. This group confirmed the presence of the ryanodine receptor in the IMM using [3H]ryanodine binding, RyR antibody conjugated immunogold particles, and Western blot analysis[85] [92]. It could serve as an alternative mechanism for Ca2+ accumulation in mitochondria as well as a regulator of Ca2+ efflux under mitochondrial Ca2+overload and pathological conditions[85][86][87] [92–94]. Interestingly, the single channel activity of mRyR was confirmed on recombinant mRyR proteins reconstituted in supported lipid bilayers prepared from IMM vesicles[88] [95]. This study elucidates pharmacological and electrophysiological features of mRyR in the model of IMM merged to lipid bilayers, where a mitochondrial transporter with gating properties similar to those of RyR in ER/SR was demonstrated[88] [95].
2.2.4 The mechanism of Ca2+ uptake including LETM1
LETM1 (leucine zipper- EF-hand containing transmembrane protein, 70 kDa) is an integral mitochondrial inner membrane protein, usually co-localized with a mitochondrial matrix protein HSP60[57][89] [62,96]. The N-terminus of this protein is linked to the IMM via a transmembrane domain consisting of 3 proline residues; whereas the C-terminus extends to the mitochondrial matrix[57][58][90] [62,63,97]. It was also demonstrated previously that LETM1 is an endogenous protein in HeLa cells, with a molecular weight of 83 kDa, and it has been assumed that it is initially produced as a cytosolic precursor with a presequence[57][89][91][92] [62,96,98,99]. LETM1 is a transporter protein shown to exhibit Ca2+/H⁺ exchange activity, acting as a crucial component in the regulation of Ca2+ homeostasis[57][89][93][94][95][96][97] [62,96,100–104]. Later it was proposed as an inner mitochondrial membrane Ca2+/H+ antiporter[96] [103] that is able to transport Ca2+ bidirectionally across the membrane. In addition, experimental work indicated the important role of LETM1 in maintaining K+ homeostasis, and this has led to the suggestion that LETM1 works as an H+/K+ exchanger with an electroneutral activity (1H+/1K+)[98] [105]. Of note, this exchanger shares a key role with MCU to catalyze Ruthenium Red-sensitive transport of Ca2+ into mitochondria[96] [103]. It would likely serve as an alternative mechanism for Ca2+ accumulation in mitochondria, as well as a regulator of Ca2+ efflux under mitochondrial Ca2+ overload[56][94][99] [61,101,106]. In summary, although the importance of LETM1 for cellular functioning is clear, the molecular characteristics and details of LETM1 organization still remain unclear.
2.3 Calcium efflux through IMM
In order to maintain the intra-mitochondrial Ca2+ homeostasis under physiological and pathological conditions, the balance between Ca2+ influx and efflux into/from mitochondria has to be maintained. The functional and molecular characterization of the mitochondrial Ca2+ efflux system already had started in the 1970s, when Na+-dependent Ca2+ efflux from mitochondria was described in isolated rat heart mitochondria[100] [107], and two different mechanisms were proposed: (1) Na+-dependent (Na+/Ca2+/Li+ exchange, NCLX) and (2) Na+-independent (H+/Ca2+ exchange, HCX) mechanisms. It was reported that the Na+/Ca2+ exchange takes place in excitable tissues (i.e., brain, heart); whereas H+/Ca2+ exchange is typical for non-excitable tissues (i.e., liver). However, both systems provide slow Ca2+ release in comparison to the rate of Ca2+ influx through the MCU [108,109]. Later, (3) the mitochondrial permeability transition pore complex (mPTPC) was identified as an important Ca2+ efflux mechanism [101][26][3,110]. Besides this, LETM1 (4) has been proposed as an additional Ca2+ efflux system[2][96][101][102] [12,103,111,112] (Figure 1).
2.3.1 The mechanism of Ca2+ efflux by NCLX
NCLX (Na+/Ca2+/Li+ exchanger systems): Mitochondrial Na+/Ca2+ (NCX) exchange was discovered by Carafoli et al. in 1974[100] [107]. However, the molecular composition of the Na+-dependent Ca2+ efflux system was resolved relatively recently[103] [113], and interestingly, seems to function as a transporter of Li+ ions as well, being a member of the family of Na+/Ca2+ exchangers[66][103][104][105] [73,113–115]. The ability of NCLX to conduct both Na+/Ca2+ and Li+/Ca2+ transport is a unique feature of the mitochondrial carrier[66][105][106] [73,115,116]. In fact, it can transport either Li+ or Na+ in exchange for Ca2+. NCLX is the only known member of the Na+/Ca2+ exchanger superfamily that can also transport Li+[66][105] [73,115]. Na+/Ca2+ exchangers are characterized as transporters with a low affinity and high capacity; thus they could be most effective in regulating of Ca2+ homeostasis during transient Ca2+ fluxes commonly expressed in excitable cells[106][107] [116,117].
NCLX mechanism predominates in the mitochondria of cardiomyocytes, neurons, cells of the skeletal muscle, parotid gland, adrenal cortex, and brown fat[105][108][109] [115,118,119] and to a lesser extent also being present in lung mitochondria and mitochondria of the kidney and liver[20][60][110] [2,67,120]. NCLX can be inhibited by benzodiazepines and CGP37157 inhibitor of the mitochondrial Na+/Ca2+ exchanger[111][112] [121,122]. Of note, under conditions when mitochondria are depolarized, all types of Ca2+ exchangers can act in the reverse mode, pumping Ca2+ into the mitochondria[55][113] [13,123].
2.3.2 The mechanism of Ca2+ efflux by HCX
HCX (H+/Ca2+ exchanger): Na+-independent Ca2+ efflux (HCX) is prevalent in mitochondria of non-excitable cells (i.e., liver, kidney, lung, smooth muscles), in contrast to the NCLX mechanism[3][60][87][109][114][115] [16,67,94,119,124,125]. The molecular composition of the HCX is still unclear and the literature on this complex sparse; however, it is assumed to be electroneutral with the stoichiometry of 2 molecules of H+ per 1 molecule of Ca2+[115][116] [125,126]. The rate of Ca2+ efflux through HCX decreases with an increase in the pH gradient[114][116] [124,126].
2.3.3 The mechanism of Ca2+ efflux by LETM1
LETM1 (leucine zipper- EF-hand containing transmembrane protein, 70 kDa): In comparison to NCX, NCLX, or HCX, Ca2+ efflux via LETM1 does not represent the major pathway, but it could serve as an alternative mechanism for the release of Ca2+[20][2][117][102][109] [2,12,103,111,112,119]. Moreover, the activity of this protein might be essential for maintenance of the tubular shape of mitochondria and for cristae organization[7][89] [9[96]. In addition, LETM1 can work as a Ca2+/H+ antiporter (see section 2.2.4 - Mechanism of Ca2+ uptake including LETM1)[118] [127].
2.3.4 The mechanism of Ca2+ efflux by mPTP/mPTPC
mPTP/mPTPC (mitochondrial permeability transition pore or mPTP complex): mPTP or mPTPC is considered as the main transport system for Ca2+ efflux from mitochondria under pathophysiological conditions[20][26][2][21][22][119][120][121][122][123][124] [2,3,12,31,32,128–134]. Although the mPTPC was initially described in swelling experiments using the fraction of isolated mitochondria and characterized as a non-selective channel that transports ionic and nonionic molecules as early as 1979[101] [110], yet the transport mechanism of this channel actually remains poorly understood. [125][126][127][128][129][130][131][132][133][134][135][136][137][138][139][140][141][142][143][144][145][146]
It is commonly believed that mPTPC is a multi-protein system in the OMM and IMM. Originally, only regulatory components were identified. The first unambiguously established component was CypD (Fig. 1), which still remains the only protein whose involvement in mPTPC pore formation and activity regulation is undisputed[147][148][149][150][151] [135–139]. CypD can stimulate structural rearrangements in the proteins responsible for the formation of mPTPC pore channel, preventing mPTP-mediated necrosis[55][148] [13,136]. Most of the studies on the role of CypD in the regulation of mPTP relied on pharmacological cyclosporin A or transient siRNA inhibition of CypD, as well as on the results obtained on models of the knockout mouse, which demonstrated its interconnection with mPTPC [147][149][150][152][135,137,138,140].
Adenine nucleotide translocase (ANT) was initially believed to represent the main regulatory component of mPTPC [153][141]. Recent studies characterized ANT as a pore-forming component and proposed a “multi-pore model” with two separate pore-forming molecular components: one of which is ANT and the other depends on CypD[154] [142]. It is also possible that CypD and ANT function in a “dual regulatory model”, where mPTPC is regulated by both ANT and CypD[154] [142]. Moreover, it is currently believed that ANTs are multifunctional proteins, which represent not only the pore-forming component of the mPTPC but may also be crucial for mitochondrial uncoupling and for the stimulation of mitophagy[155] [143].
Furthermore, F0F1 ATP Synthase and the phosphate carrier (PiC) are considered as the core pore-forming components of mPTPC[120][156][125][126][127][128] [130,144–148]. FoF1 ATP Synthase forms the channel in mPTC and transports molecules through the 2 ATP synthase monomers or through the ring of the c-subunit, which overlaps with the IMM and the pore forming component[128][129][130] [148,149]. However, it should be noted that classification of the last named component (PiC) is more complicated, since in the context of its ability to activate mPTP opening it can be considered as the pore forming component[130] [150]. At the same time, following patch clamping of the PiC displayed too low of a conductance to assume that it functions as the core pore-forming constituent of the mPTPC. Undoubtedly, the precise nature and molecular organization of the pore-forming part of mPTPC remain controversial[120][128][129][131][132][133][134][135][136][137][138][139][140][141] [130,148,149,151–161]. m-AAA protease Spastic Paraplegia 7 (SPG7) was previously thought to be a core component of the mPTP that is able to interact with CypD and with VDAC1 at the OMM/IMM contact sites[148] [136]. However, recent results demonstrate that SPG7 is not a core component of the mPTP, but could regulate the mPTP activity by decreasing Ca2+ levels in mitochondrial matrix through modulation of MCUR1 and MCU assembly[126] [146].
The efflux of Ca2+ occurs through a transient or low conductance opening of mPTP, most likely by lower oligomeric states of mPTP[55][21][142][143][144][145][146][157][158] [13,131,162–168]. The evidence for transient opening of mPTP for Ca2+ was demonstrated by the early studies on the inhibition of Ca2+ release by Cyclosporin A in isolated adult rat ventricular cardiomyocytes[123] [133]. Transient opening or low conductance opening of the mPTP represent a Ca2+ efflux mechanism, and various studies have confirmed the essential role of mPTP in the release of Ca2+[55][121][144][145][146][157][158] [13,131,164–168]. mPTP is a nonspecific channel, used by cells in signal transduction and the transfer of molecules between the mitochondrial matrix and cytoplasm. In particular it maintains Ca2+ homeostasis, regulates oxidative stress signals, and mediates cell death[119][121][159][160] [128,131,169,170]. Regarding the multi-conductance function of mPTPC, it likely can be assumed that mPTPC is partially oligomerized into a complex with multiple subunits[122][160][161] [132,170,171]. The first studies using different sized polyethylene glycols identified solutes of up to 1500 Da that could be transported through the pore that matches the modeled pore size of 1.4 nm[101] [110]. Importantly, mPTP is able to reversibly open upon an increase in ADP concentration, as well as during restoration of the Mg2+/Ca2+ratio[101] [110], reestablishing mitochondrial membrane potential, and allowing for mPTP to have either a sustained or transient opening[122][162] [132,172]. The different regimes of mPTP opening determine the selectivity in signaling.[159][163][164][165][166][167][168][169][170][171]
The opening of mPTP is directly regulated by the concentration of free Ca2+, and triggered by mitochondrial Ca2+ overload; allowing for rapid Ca2+ release from mitochondria[26][55][22][63][122][136][172] [3,13,32,70,132,156,173]. Obviously, Ca2+ is the most important regulator and inductor of mPTP opening, regarding its numerous indirect roles in the regulation and modulation of the mPTP[120][121][122][173] [130–132,174]. The functional dualism of Ca2+ is an important factor of mPTP mediation. At physiological levels of Ca2+ it can activate transient opening of the pore; whereas at Ca2+ overload it can induce pathological changes, resulting in sustained mPTP opening and subsequent mitochondrial and cellular dysfunction [119][122][124][174][128,132,134,173,175].
Activation of mPTP could also be mediated at different levels through regulation by kinases, as well as posttranslational modification of CypD[163] [176]. It has been shown, that mPTP could be stimulated by Ca2+ in combination with an increase in the concentration of ROS and phosphate; aditionally, that it could be inhibited by divalent cations (such as Mg2+, Mn2+), adenine nucleotides, low pH, or CypD inhibitors (such as CsA and sanglifehrin A)[164] [177]. Importantly, modifications and loss of CypD induce a significant increase in the threshold concentration of Ca2+ required for pore opening[55][148] [13,136].
Hypothetically, VDAC could also mediate mPTPC activity; however, genetic analysis did not prove to be any essential function of this protein in mPTP-mediated cell death[47][174] [53,175]. Electrophysiological and biochemical studies supported the molecular model of mPTPC with the VDAC on the OMM, ANT on the IMM, and CypD in the matrix [165][166][167][178–180]. In brief, the following facts speak for involvement of VDAC1 in mPTP opening and function: overexpression of microRNA-7 prevents opening of mPTP by downregulating VDAC1 [181[168]]; the loss of mitochondrial fission factor Mff inhibits mPTP opening via blocking of VDAC1 oligomerization and separation of HKII, which leads to the inhibition of mPTP opening [169][1[182]. On the other hand, additional studies have provided opposing results, indicating that the closed state of VDAC stimulates Ca2+ permeability, and therefore forces mPTP opening [183,184].
[1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102][103][104][105][106][107][108][109][110][111][112][113][114][115][116][117][118][119][120][121][122][123][124][125][126][127][128][129][130][131][132][133][134][135][136][137][138][139][140][141][142][143][144][145][146][147][148][149][150][151][152][153][154][155][156][157][158][159][160][161][162][163][164][165][166][167][168][169][170][171][172][173][174][175].
References
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814, doi:10.1007/s00018-013-1550-7.Carafoli, E.; Krebs, J. Why Calcium? How Calcium Became the Best Communicator. J. Biol. Chem. 2016, 291, 20849–20857, doi:10.1074/jbc.R116.735894.
- Delierneux, C.; Kouba, S.; Shanmughapriya, S.; Potier-Cartereau, M.; Trebak, M.; Hempel, N. Mitochondrial Calcium Regulation of Redox Signaling in Cancer. Cells 2020, 9, doi:10.3390/cells9020432.Membrane Dynamics and Calcium Signaling; Krebs, J., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, 2017; Vol. 981; ISBN 978-3-319-55857-8.
- McCarron, J.G.; Saunter, C.; Wilson, C.; Girkin, J.M.; Chalmers, S. Mitochondria Structure and Position in the Local Control of Calcium Signals in Smooth Muscle Cells; CRC Press, 2018; ISBN 9781498774222.Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol. Rev. 2019, 99, 1765–1817, doi:10.1152/physrev.00022.2018.
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100, doi:10.1038/s41580-019-0173-8.Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195, doi:10.1161/CIRCRESAHA.117.310230.
- Wacquier, B.; Combettes, L.; Dupont, G. Dual dynamics of mitochondrial permeability transition pore opening. Sci. Rep. 2020, 10, 3924, doi:10.1038/s41598-020-60177-1.Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814, doi:10.1007/s00018-013-1550-7.
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta - Bioenerg. 2009, 1787, 1309–1316, doi:10.1016/j.bbabio.2009.01.005.Duchen, M.R. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem. J. 1992, 283, 41–50, doi:10.1042/bj2830041.
- Mitochondrial Dynamics in Cardiovascular Medicine; Santulli, G., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, 2017; Vol. 982; ISBN 978-3-319-55329-0.Elustondo, P.A.; Nichols, M.; Robertson, G.S.; Pavlov, E. V Mitochondrial Ca2+ uptake pathways. J. Bioenerg. Biomembr. 2017, 49, 113—119, doi:10.1007/s10863-016-9676-6.
- Picard, M.; Wallace, D.C.; Burelle, Y. The rise of mitochondria in medicine. Mitochondrion 2016, 30, 105–116, doi:10.1016/j.mito.2016.07.003.Duchen, M.R. Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol. Aspects Med. 2004, 25, 365–451, doi:10.1016/j.mam.2004.03.001.
- Duchen, M.R. Mitochondria and calcium: from cell signalling to cell death. J. Physiol. 2000, 529, 57–68, doi:10.1111/j.1469-7793.2000.00057.x.Mitochondrial Dynamics in Cardiovascular Medicine; Santulli, G., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, 2017; Vol. 982; ISBN 978-3-319-55329-0.
- Park, M.K. Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J. 2001, 20, 1863–1874, doi:10.1093/emboj/20.8.1863.Bravo-Sagua, R.; Parra, V.; Lopez-Crisosto, C.; Diaz, P.; Quest, A.F.G.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634, doi:10.1002/cphy.c160013.
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730, doi:10.1038/s41580-018-0052-8.Giorgio, V.; Guo, L.; Bassot, C.; Petronilli, V.; Bernardi, P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 2018, 70, 56–63, doi:10.1016/j.ceca.2017.05.004.
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224, doi:10.1038/s41580-020-0210-7.Delierneux, C.; Kouba, S.; Shanmughapriya, S.; Potier-Cartereau, M.; Trebak, M.; Hempel, N. Mitochondrial Calcium Regulation of Redox Signaling in Cancer. Cells 2020, 9, doi:10.3390/cells9020432.
- Morciano, G.; Marchi, S.; Morganti, C.; Sbano, L.; Bittremieux, M.; Kerkhofs, M.; Corricelli, M.; Danese, A.; Karkucinska-Wieckowska, A.; Wieckowski, M.R.; et al. Role of Mitochondria-Associated ER Membranes in Calcium Regulation in Cancer-Specific Settings. Neoplasia 2018, 20, 510–523, doi:10.1016/j.neo.2018.03.005.Belosludtsev, K.N.; Dubinin, M. V; Belosludtseva, N. V; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry. (Mosc). 2019, 84, 593–607, doi:10.1134/S0006297919060026.
- Herrera-Cruz, M.S.; Simmen, T. Cancer: Untethering Mitochondria from the Endoplasmic Reticulum? Front. Oncol. 2017, 7, doi:10.3389/fonc.2017.00105.Hausenloy, D.J.; Schulz, R.; Girao, H.; Kwak, B.R.; De Stefani, D.; Rizzuto, R.; Bernardi, P.; Di Lisa, F. Mitochondrial ion channels as targets for cardioprotection. J. Cell. Mol. Med. 2020, 24, 7102–7114, doi:10.1111/jcmm.15341.
- Singaravelu, K.; Nelson, C.; Bakowski, D.; de Brito, O.M.; Ng, S.-W.; Di Capite, J.; Powell, T.; Scorrano, L.; Parekh, A.B. Mitofusin 2 Regulates STIM1 Migration from the Ca 2+ Store to the Plasma Membrane in Cells with Depolarized Mitochondria. J. Biol. Chem. 2011, 286, 12189–12201, doi:10.1074/jbc.M110.174029.Glaser, T.; Arnaud Sampaio, V.F.; Lameu, C.; Ulrich, H. Calcium signalling: A common target in neurological disorders and neurogenesis. Semin. Cell Dev. Biol. 2019, 95, 25–33, doi:10.1016/j.semcdb.2018.12.002.
- Duchen, M.R. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem. J. 1992, 283, 41–50, doi:10.1042/bj2830041.McCarron, J.G.; Saunter, C.; Wilson, C.; Girkin, J.M.; Chalmers, S. Mitochondria Structure and Position in the Local Control of Calcium Signals in Smooth Muscle Cells; CRC Press, 2018; ISBN 9781498774222.
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. 1999, 96, 13807–13812, doi:10.1073/pnas.96.24.13807.Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100, doi:10.1038/s41580-019-0173-8.
- Carpio, M.A.; Katz, S.G. Methods to Probe Calcium Regulation by BCL-2 Family Members. Methods Mol. Biol. 2019, 1877, 173–183, doi:10.1007/978-1-4939-8861-7_12.Wacquier, B.; Combettes, L.; Dupont, G. Dual dynamics of mitochondrial permeability transition pore opening. Sci. Rep. 2020, 10, 3924, doi:10.1038/s41598-020-60177-1.
- Rong, Y.; Distelhorst, C.W. Bcl-2 Protein Family Members: Versatile Regulators of Calcium Signaling in Cell Survival and Apoptosis. Annu. Rev. Physiol. 2008, 70, 73–91, doi:10.1146/annurev.physiol.70.021507.105852.Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta - Bioenerg. 2009, 1787, 1309–1316, doi:10.1016/j.bbabio.2009.01.005.
- Membrane Dynamics and Calcium Signaling; Krebs, J., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, 2017; Vol. 981; ISBN 978-3-319-55857-8.Picard, M.; Wallace, D.C.; Burelle, Y. The rise of mitochondria in medicine. Mitochondrion 2016, 30, 105–116, doi:10.1016/j.mito.2016.07.003.
- Briston, T.; Roberts, M.; Lewis, S.; Powney, B.; M. Staddon, J.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition pore: sensitivity to opening and mechanistic dependence on substrate availability. Sci. Rep. 2017, 7, 10492, doi:10.1038/s41598-017-10673-8.Duchen, M.R. Mitochondria and calcium: from cell signalling to cell death. J. Physiol. 2000, 529, 57–68, doi:10.1111/j.1469-7793.2000.00057.x.
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules 2020, 10, 998, doi:10.3390/biom10070998.Park, M.K. Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J. 2001, 20, 1863–1874, doi:10.1093/emboj/20.8.1863.
- VASINGTON, F.D.; MURPHY, J. V Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J. Biol. Chem. 1962, 237, 2670–2677.Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730, doi:10.1038/s41580-018-0052-8.
- DeLuca, H.F.; Engstrom, G.W. CALCIUM UPTAKE BY RAT KIDNEY MITOCHONDRIA. Proc. Natl. Acad. Sci. 1961, 47, 1744–1750, doi:10.1073/pnas.47.11.1744.Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224, doi:10.1038/s41580-020-0210-7.
- MITCHELL, P.; MOYLE, J. Chemiosmotic Hypothesis of Oxidative Phosphorylation. Nature 1967, 213, 137–139, doi:10.1038/213137a0.Morciano, G.; Marchi, S.; Morganti, C.; Sbano, L.; Bittremieux, M.; Kerkhofs, M.; Corricelli, M.; Danese, A.; Karkucinska-Wieckowska, A.; Wieckowski, M.R.; et al. Role of Mitochondria-Associated ER Membranes in Calcium Regulation in Cancer-Specific Settings. Neoplasia 2018, 20, 510–523, doi:10.1016/j.neo.2018.03.005.
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol. Rev. 2019, 99, 1765–1817, doi:10.1152/physrev.00022.2018.Herrera-Cruz, M.S.; Simmen, T. Cancer: Untethering Mitochondria from the Endoplasmic Reticulum? Front. Oncol. 2017, 7, doi:10.3389/fonc.2017.00105.
- Elustondo, P.A.; Nichols, M.; Robertson, G.S.; Pavlov, E. V Mitochondrial Ca2+ uptake pathways. J. Bioenerg. Biomembr. 2017, 49, 113—119, doi:10.1007/s10863-016-9676-6.Singaravelu, K.; Nelson, C.; Bakowski, D.; de Brito, O.M.; Ng, S.-W.; Di Capite, J.; Powell, T.; Scorrano, L.; Parekh, A.B. Mitofusin 2 Regulates STIM1 Migration from the Ca 2+ Store to the Plasma Membrane in Cells with Depolarized Mitochondria. J. Biol. Chem. 2011, 286, 12189–12201, doi:10.1074/jbc.M110.174029.
- Bhosale, G.; Sharpe, J.A.; Sundier, S.Y.; Duchen, M.R. Calcium signaling as a mediator of cell energy demand and a trigger to cell death. Ann. N. Y. Acad. Sci. 2015, 1350, 107–116, doi:10.1111/nyas.12885.Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. 1999, 96, 13807–13812, doi:10.1073/pnas.96.24.13807.
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. acta. Mol. cell Res. 2019, 1866, 1068–1078, doi:10.1016/j.bbamcr.2018.10.016.Carpio, M.A.; Katz, S.G. Methods to Probe Calcium Regulation by BCL-2 Family Members. Methods Mol. Biol. 2019, 1877, 173–183, doi:10.1007/978-1-4939-8861-7_12.
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478, doi:10.1161/CIRCRESAHA.118.310082.Rong, Y.; Distelhorst, C.W. Bcl-2 Protein Family Members: Versatile Regulators of Calcium Signaling in Cell Survival and Apoptosis. Annu. Rev. Physiol. 2008, 70, 73–91, doi:10.1146/annurev.physiol.70.021507.105852.
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. EBioMedicine 2020, 59, 102943, doi:10.1016/j.ebiom.2020.102943.Briston, T.; Roberts, M.; Lewis, S.; Powney, B.; M. Staddon, J.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition pore: sensitivity to opening and mechanistic dependence on substrate availability. Sci. Rep. 2017, 7, 10492, doi:10.1038/s41598-017-10673-8.
- Hausenloy, D.J.; Schulz, R.; Girao, H.; Kwak, B.R.; De Stefani, D.; Rizzuto, R.; Bernardi, P.; Di Lisa, F. Mitochondrial ion channels as targets for cardioprotection. J. Cell. Mol. Med. 2020, 24, 7102–7114, doi:10.1111/jcmm.15341.Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules 2020, 10, 998, doi:10.3390/biom10070998.
- Glaser, T.; Arnaud Sampaio, V.F.; Lameu, C.; Ulrich, H. Calcium signalling: A common target in neurological disorders and neurogenesis. Semin. Cell Dev. Biol. 2019, 95, 25–33, doi:10.1016/j.semcdb.2018.12.002.VASINGTON, F.D.; MURPHY, J. V Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J. Biol. Chem. 1962, 237, 2670–2677.
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox Signaling in Cardiac Physiology and Pathology. Circ. Res. 2012, 111, 1091–1106, doi:10.1161/CIRCRESAHA.111.255216.DeLuca, H.F.; Engstrom, G.W. CALCIUM UPTAKE BY RAT KIDNEY MITOCHONDRIA. Proc. Natl. Acad. Sci. 1961, 47, 1744–1750, doi:10.1073/pnas.47.11.1744.
- Bravo-Sagua, R.; Parra, V.; Lopez-Crisosto, C.; Diaz, P.; Quest, A.F.G.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634, doi:10.1002/cphy.c160013.MITCHELL, P.; MOYLE, J. Chemiosmotic Hypothesis of Oxidative Phosphorylation. Nature 1967, 213, 137–139, doi:10.1038/213137a0.
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: from molecular mechanism to animal models. Pflugers Arch. 2018, 470, 1165–1179, doi:10.1007/s00424-018-2123-2.Ludtmann, M.H.R.; Abramov, A.Y. Mitochondrial calcium imbalance in Parkinson’s disease. Neurosci. Lett. 2018, 663, 86–90, doi:10.1016/j.neulet.2017.08.044.
- Ben-Hail, D.; Shoshan-Barmatz, V. VDAC1-interacting anion transport inhibitors inhibit VDAC1 oligomerization and apoptosis. Biochim. Biophys. Acta - Mol. Cell Res. 2016, 1863, 1612–1623, doi:10.1016/j.bbamcr.2016.04.002.Bhosale, G.; Sharpe, J.A.; Sundier, S.Y.; Duchen, M.R. Calcium signaling as a mediator of cell energy demand and a trigger to cell death. Ann. N. Y. Acad. Sci. 2015, 1350, 107–116, doi:10.1111/nyas.12885.
- Schein, S.J.; Colombini, M.; Finkelstein, A. Reconstitution in planar lipid bilayers of a voltage-dependent anion-selective channel obtained from paramecium mitochondria. J. Membr. Biol. 1976, 30, 99–120, doi:10.1007/bf01869662.Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. acta. Mol. cell Res. 2019, 1866, 1068–1078, doi:10.1016/j.bbamcr.2018.10.016.
- Mazure, N.M. VDAC in cancer. Biochim. Biophys. Acta - Bioenerg. 2017, 1858, 665–673, doi:10.1016/j.bbabio.2017.03.002.Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478, doi:10.1161/CIRCRESAHA.118.310082.
- Becker, T.; Wagner, R. Mitochondrial Outer Membrane Channels: Emerging Diversity in Transport Processes. BioEssays 2018, 40, 1800013, doi:10.1002/bies.201800013.Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. EBioMedicine 2020, 59, 102943, doi:10.1016/j.ebiom.2020.102943.
- Rostovtseva, T.K.; Queralt-Martín, M.; Rosencrans, W.M.; Bezrukov, S.M. Targeting the Multiple Physiologic Roles of VDAC With Steroids and Hydrophobic Drugs. Front. Physiol. 2020, 11, 446, doi:10.3389/fphys.2020.00446.Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox Signaling in Cardiac Physiology and Pathology. Circ. Res. 2012, 111, 1091–1106, doi:10.1161/CIRCRESAHA.111.255216.
- Shoshan-Barmatz, V.; Gincel, D. The voltage-dependent anion channel: characterization, modulation, and role in mitochondrial function in cell life and death. Cell Biochem. Biophys. 2003, 39, 279–292, doi:10.1385/CBB:39:3:279.Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: from molecular mechanism to animal models. Pflugers Arch. 2018, 470, 1165–1179, doi:10.1007/s00424-018-2123-2.
- Colombini, M.; Mannella, C.A. VDAC, The early days. Biochim. Biophys. Acta - Biomembr. 2012, 1818, 1438–1443, doi:10.1016/j.bbamem.2011.11.014.Ben-Hail, D.; Shoshan-Barmatz, V. VDAC1-interacting anion transport inhibitors inhibit VDAC1 oligomerization and apoptosis. Biochim. Biophys. Acta - Mol. Cell Res. 2016, 1863, 1612–1623, doi:10.1016/j.bbamcr.2016.04.002.
- COLOMBINI, M. A candidate for the permeability pathway of the outer mitochondrial membrane. Nature 1979, 279, 643–645, doi:10.1038/279643a0.Schein, S.J.; Colombini, M.; Finkelstein, A. Reconstitution in planar lipid bilayers of a voltage-dependent anion-selective channel obtained from paramecium mitochondria. J. Membr. Biol. 1976, 30, 99–120, doi:10.1007/bf01869662.
- Kusano, T.; Tateda, C.; Berberich, T.; Takahashi, Y. Voltage-dependent anion channels: their roles in plant defense and cell death. Plant Cell Rep. 2009, 28, 1301–1308, doi:10.1007/s00299-009-0741-z.Mazure, N.M. VDAC in cancer. Biochim. Biophys. Acta - Bioenerg. 2017, 1858, 665–673, doi:10.1016/j.bbabio.2017.03.002.
- Shoshan-Barmatz, V.; Mizrachi, D. VDAC1: from structure to cancer therapy. Front. Oncol. 2012, 2, 164, doi:10.3389/fonc.2012.00164.Becker, T.; Wagner, R. Mitochondrial Outer Membrane Channels: Emerging Diversity in Transport Processes. BioEssays 2018, 40, 1800013, doi:10.1002/bies.201800013.
- Camara, A.K.S.; Zhou, Y.; Wen, P.-C.; Tajkhorshid, E.; Kwok, W.-M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, doi:10.3389/fphys.2017.00460.Rostovtseva, T.K.; Queralt-Martín, M.; Rosencrans, W.M.; Bezrukov, S.M. Targeting the Multiple Physiologic Roles of VDAC With Steroids and Hydrophobic Drugs. Front. Physiol. 2020, 11, 446, doi:10.3389/fphys.2020.00446.
- Ponnalagu, D.; Singh, H. Anion Channels of Mitochondria. In Handbook of experimental pharmacology; Germany, 2016; Vol. 240, pp. 71–101.Shoshan-Barmatz, V.; Gincel, D. The voltage-dependent anion channel: characterization, modulation, and role in mitochondrial function in cell life and death. Cell Biochem. Biophys. 2003, 39, 279–292, doi:10.1385/CBB:39:3:279.
- Neumann, D.; Bückers, J.; Kastrup, L.; Hell, S.W.; Jakobs, S. Two-color STED microscopy reveals different degrees of colocalization between hexokinase-I and the three human VDAC isoforms. PMC Biophys. 2010, 3, 4, doi:10.1186/1757-5036-3-4.Colombini, M.; Mannella, C.A. VDAC, The early days. Biochim. Biophys. Acta - Biomembr. 2012, 1818, 1438–1443, doi:10.1016/j.bbamem.2011.11.014.
- Geula, S.; Ben-Hail, D.; Shoshan-Barmatz, V. Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem. J. 2012, 444, 475–485, doi:10.1042/BJ20112079.COLOMBINI, M. A candidate for the permeability pathway of the outer mitochondrial membrane. Nature 1979, 279, 643–645, doi:10.1038/279643a0.
- Cheng, E.H.Y.; Sheiko, T. V; Fisher, J.K.; Craigen, W.J.; Korsmeyer, S.J. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 2003, 301, 513–517, doi:10.1126/science.1083995.Kusano, T.; Tateda, C.; Berberich, T.; Takahashi, Y. Voltage-dependent anion channels: their roles in plant defense and cell death. Plant Cell Rep. 2009, 28, 1301–1308, doi:10.1007/s00299-009-0741-z.
- Checchetto, V.; Reina, S.; Magrì, A.; Szabo, I.; De Pinto, V. Recombinant Human Voltage Dependent Anion Selective Channel Isoform 3 (hVDAC3) Forms Pores with a Very Small Conductance. Cell. Physiol. Biochem. 2014, 34, 842–853, doi:10.1159/000363047.Shoshan-Barmatz, V.; Mizrachi, D. VDAC1: from structure to cancer therapy. Front. Oncol. 2012, 2, 164, doi:10.3389/fonc.2012.00164.
- De Pinto, V.; Guarino, F.; Guarnera, A.; Messina, A.; Reina, S.; Tomasello, F.M.; Palermo, V.; Mazzoni, C. Characterization of human VDAC isoforms: a peculiar function for VDAC3? Biochim. Biophys. Acta 2010, 1797, 1268—1275, doi:10.1016/j.bbabio.2010.01.031.Camara, A.K.S.; Zhou, Y.; Wen, P.-C.; Tajkhorshid, E.; Kwok, W.-M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, doi:10.3389/fphys.2017.00460.
- Lemasters, J.J.; Holmuhamedov, E.L.; Czerny, C.; Zhong, Z.; Maldonado, E.N. Regulation of mitochondrial function by voltage dependent anion channels in ethanol metabolism and the Warburg effect. Biochim. Biophys. Acta 2012, 1818, 1536—1544, doi:10.1016/j.bbamem.2011.11.034.Ponnalagu, D.; Singh, H. Anion Channels of Mitochondria. In Handbook of experimental pharmacology; Germany, 2016; Vol. 240, pp. 71–101.
- Belosludtsev, K.N.; Dubinin, M. V; Belosludtseva, N. V; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry. (Mosc). 2019, 84, 593–607, doi:10.1134/S0006297919060026.Neumann, D.; Bückers, J.; Kastrup, L.; Hell, S.W.; Jakobs, S. Two-color STED microscopy reveals different degrees of colocalization between hexokinase-I and the three human VDAC isoforms. PMC Biophys. 2010, 3, 4, doi:10.1186/1757-5036-3-4.
- Austin, S.; Nowikovsky, K. LETM1: Essential for Mitochondrial Biology and Cation Homeostasis? Trends Biochem. Sci. 2019, 44, 648–658, doi:10.1016/j.tibs.2019.04.002.Geula, S.; Ben-Hail, D.; Shoshan-Barmatz, V. Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem. J. 2012, 444, 475–485, doi:10.1042/BJ20112079.
- Li, Y.; Tran, Q.; Shrestha, R.; Piao, L.; Park, S.; Park, J.; Park, J. LETM1 is required for mitochondrial homeostasis and cellular viability (Review). Mol. Med. Rep. 2019, 19, 3367–3375, doi:10.3892/mmr.2019.10041.Cheng, E.H.Y.; Sheiko, T. V; Fisher, J.K.; Craigen, W.J.; Korsmeyer, S.J. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 2003, 301, 513–517, doi:10.1126/science.1083995.
- Shao, J.; Fu, Z.; Ji, Y.; Guan, X.; Guo, S.; Ding, Z.; Yang, X.; Cong, Y.; Shen, Y. Leucine zipper-EF-hand containing transmembrane protein 1 (LETM1) forms a Ca(2+)/H(+) antiporter. Sci. Rep. 2016, 6, 34174, doi:10.1038/srep34174.Checchetto, V.; Reina, S.; Magrì, A.; Szabo, I.; De Pinto, V. Recombinant Human Voltage Dependent Anion Selective Channel Isoform 3 (hVDAC3) Forms Pores with a Very Small Conductance. Cell. Physiol. Biochem. 2014, 34, 842–853, doi:10.1159/000363047.
- Waldeck-Weiermair, M.; Jean-Quartier, C.; Rost, R.; Khan, M.J.; Vishnu, N.; Bondarenko, A.I.; Imamura, H.; Malli, R.; Graier, W.F. Leucine Zipper EF Hand-containing Transmembrane Protein 1 (Letm1) and Uncoupling Proteins 2 and 3 (UCP2/3) Contribute to Two Distinct Mitochondrial Ca 2+ Uptake Pathways. J. Biol. Chem. 2011, 286, 28444–28455, doi:10.1074/jbc.M111.244517.De Pinto, V.; Guarino, F.; Guarnera, A.; Messina, A.; Reina, S.; Tomasello, F.M.; Palermo, V.; Mazzoni, C. Characterization of human VDAC isoforms: a peculiar function for VDAC3? Biochim. Biophys. Acta 2010, 1797, 1268—1275, doi:10.1016/j.bbabio.2010.01.031.
- Gunter, T.E.; Pfeiffer, D.R. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 1990, 258, C755-86, doi:10.1152/ajpcell.1990.258.5.C755.Lemasters, J.J.; Holmuhamedov, E.L.; Czerny, C.; Zhong, Z.; Maldonado, E.N. Regulation of mitochondrial function by voltage dependent anion channels in ethanol metabolism and the Warburg effect. Biochim. Biophys. Acta 2012, 1818, 1536—1544, doi:10.1016/j.bbamem.2011.11.034.
- Mishra, J.; Jhun, B.S.; Hurst, S.; O-Uchi, J.; Csordás, G.; Sheu, S.-S. The Mitochondrial Ca2+ Uniporter: Structure, Function, and Pharmacology. Handb. Exp. Pharmacol. 2017, 240, 129—156, doi:10.1007/164_2017_1.Austin, S.; Nowikovsky, K. LETM1: Essential for Mitochondrial Biology and Cation Homeostasis? Trends Biochem. Sci. 2019, 44, 648–658, doi:10.1016/j.tibs.2019.04.002.
- Pallafacchina, G.; Zanin, S.; Rizzuto, R. Recent advances in the molecular mechanism of mitochondrial calcium uptake. F1000Research 2018, 7, 1858, doi:10.12688/f1000research.15723.1.Li, Y.; Tran, Q.; Shrestha, R.; Piao, L.; Park, S.; Park, J.; Park, J. LETM1 is required for mitochondrial homeostasis and cellular viability (Review). Mol. Med. Rep. 2019, 19, 3367–3375, doi:10.3892/mmr.2019.10041.
- Granatiero, V.; De Stefani, D.; Rizzuto, R. Mitochondrial Calcium Handling in Physiology and Disease. Adv. Exp. Med. Biol. 2017, 982, 25–47, doi:10.1007/978-3-319-55330-6_2.Shao, J.; Fu, Z.; Ji, Y.; Guan, X.; Guo, S.; Ding, Z.; Yang, X.; Cong, Y.; Shen, Y. Leucine zipper-EF-hand containing transmembrane protein 1 (LETM1) forms a Ca(2+)/H(+) antiporter. Sci. Rep. 2016, 6, 34174, doi:10.1038/srep34174.
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382, doi:10.1126/science.1242993.Waldeck-Weiermair, M.; Jean-Quartier, C.; Rost, R.; Khan, M.J.; Vishnu, N.; Bondarenko, A.I.; Imamura, H.; Malli, R.; Graier, W.F. Leucine Zipper EF Hand-containing Transmembrane Protein 1 (Letm1) and Uncoupling Proteins 2 and 3 (UCP2/3) Contribute to Two Distinct Mitochondrial Ca 2+ Uptake Pathways. J. Biol. Chem. 2011, 286, 28444–28455, doi:10.1074/jbc.M111.244517.
- Cui, C.; Yang, J.; Fu, L.; Wang, M.; Wang, X. Progress in understanding mitochondrial calcium uniporter complex‐mediated calcium signalling: A potential target for cancer treatment. Br. J. Pharmacol. 2019, 176, 1190–1205, doi:10.1111/bph.14632.De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340, doi:10.1038/nature10230.
- Pathak, T.; Trebak, M. Mitochondrial Ca2+ signaling. Pharmacol. Ther. 2018, 192, 112–123, doi:10.1016/j.pharmthera.2018.07.001.Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345, doi:10.1038/nature10234.
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabo, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376, doi:10.1038/emboj.2013.157.Gunter, T.E.; Pfeiffer, D.R. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 1990, 258, C755-86, doi:10.1152/ajpcell.1990.258.5.C755.
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabo, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737, doi:10.1016/j.molcel.2014.01.013.Mishra, J.; Jhun, B.S.; Hurst, S.; O-Uchi, J.; Csordás, G.; Sheu, S.-S. The Mitochondrial Ca2+ Uniporter: Structure, Function, and Pharmacology. Handb. Exp. Pharmacol. 2017, 240, 129—156, doi:10.1007/164_2017_1.
- Csordás, G.; Golenár, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; de la Fuente Perez, S.; Bogorad, R.; et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013, 17, 976—987, doi:10.1016/j.cmet.2013.04.020.Pallafacchina, G.; Zanin, S.; Rizzuto, R. Recent advances in the molecular mechanism of mitochondrial calcium uptake. F1000Research 2018, 7, 1858, doi:10.12688/f1000research.15723.1.
- Wang, L.; Yang, X.; Li, S.; Wang, Z.; Liu, Y.; Feng, J.; Zhu, Y.; Shen, Y. Structural and mechanistic insights into MICU1 regulation of mitochondrial calcium uptake. EMBO J. 2014, 33, 594–604, doi:10.1002/embj.201386523.Granatiero, V.; De Stefani, D.; Rizzuto, R. Mitochondrial Calcium Handling in Physiology and Disease. Adv. Exp. Med. Biol. 2017, 982, 25–47, doi:10.1007/978-3-319-55330-6_2.
- Vais, H.; Payne, R.; Paudel, U.; Li, C.; Foskett, J.K. Coupled transmembrane mechanisms control MCU-mediated mitochondrial Ca2+ uptake. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 21731–21739, doi:10.1073/pnas.2005976117.Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382, doi:10.1126/science.1242993.
- Paillard, M.; Csordás, G.; Szanda, G.; Golenár, T.; Debattisti, V.; Bartok, A.; Wang, N.; Moffat, C.; Seifert, E.L.; Spät, A.; et al. Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca 2+ Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU. Cell Rep. 2017, 18, 2291–2300, doi:10.1016/j.celrep.2017.02.032.Cui, C.; Yang, J.; Fu, L.; Wang, M.; Wang, X. Progress in understanding mitochondrial calcium uniporter complex‐mediated calcium signalling: A potential target for cancer treatment. Br. J. Pharmacol. 2019, 176, 1190–1205, doi:10.1111/bph.14632.
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 2013, 8, e55785, doi:10.1371/journal.pone.0055785.Pathak, T.; Trebak, M. Mitochondrial Ca2+ signaling. Pharmacol. Ther. 2018, 192, 112–123, doi:10.1016/j.pharmthera.2018.07.001.
- Kamer, K.J.; Grabarek, Z.; Mootha, V.K. High‐affinity cooperative Ca 2+ binding by MICU 1– MICU 2 serves as an on–off switch for the uniporter. EMBO Rep. 2017, 18, 1397–1411, doi:10.15252/embr.201643748.Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabo, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376, doi:10.1038/emboj.2013.157.
- Payne, R.; Hoff, H.; Roskowski, A.; Foskett, J.K. MICU2 Restricts Spatial Crosstalk between InsP 3 R and MCU Channels by Regulating Threshold and Gain of MICU1-Mediated Inhibition and Activation of MCU. Cell Rep. 2017, 21, 3141–3154, doi:10.1016/j.celrep.2017.11.064.Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabo, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737, doi:10.1016/j.molcel.2014.01.013.
- Mallilankaraman, K.; Cardenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenar, T.; Csordas, G.; Madireddi, P.; Yang, J.; Muller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343, doi:10.1038/ncb2622.Csordás, G.; Golenár, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; de la Fuente Perez, S.; Bogorad, R.; et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013, 17, 976—987, doi:10.1016/j.cmet.2013.04.020.
- Ren, T.; Wang, J.; Zhang, H.; Yuan, P.; Zhu, J.; Wu, Y.; Huang, Q.; Guo, X.; Zhang, J.; Ji, L.; et al. MCUR1-Mediated Mitochondrial Calcium Signaling Facilitates Cell Survival of Hepatocellular Carcinoma via Reactive Oxygen Species-Dependent P53 Degradation. Antioxid. Redox Signal. 2018, 28, 1120–1136, doi:10.1089/ars.2017.6990.Wang, L.; Yang, X.; Li, S.; Wang, Z.; Liu, Y.; Feng, J.; Zhu, Y.; Shen, Y. Structural and mechanistic insights into MICU1 regulation of mitochondrial calcium uptake. EMBO J. 2014, 33, 594–604, doi:10.1002/embj.201386523.
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S.J.; Breves, S.L.; Corbally, D.P.; et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016, 15, 1673–1685, doi:10.1016/j.celrep.2016.04.050.Vais, H.; Payne, R.; Paudel, U.; Li, C.; Foskett, J.K. Coupled transmembrane mechanisms control MCU-mediated mitochondrial Ca2+ uptake. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 21731–21739, doi:10.1073/pnas.2005976117.
- Paupe, V.; Prudent, J.; Dassa, E.P.; Rendon, O.Z.; Shoubridge, E.A. CCDC90A (MCUR1) Is a Cytochrome c Oxidase Assembly Factor and Not a Regulator of the Mitochondrial Calcium Uniporter. Cell Metab. 2015, 21, 109–116, doi:10.1016/j.cmet.2014.12.004.Paillard, M.; Csordás, G.; Szanda, G.; Golenár, T.; Debattisti, V.; Bartok, A.; Wang, N.; Moffat, C.; Seifert, E.L.; Spät, A.; et al. Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca 2+ Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU. Cell Rep. 2017, 18, 2291–2300, doi:10.1016/j.celrep.2017.02.032.
- Bassi, M.T.; Manzoni, M.; Bresciani, R.; Pizzo, M.T.; Della Monica, A.; Barlati, S.; Monti, E.; Borsani, G. Cellular expression and alternative splicing of SLC25A23, a member of the mitochondrial Ca2+-dependent solute carrier gene family. Gene 2005, 345, 173–182, doi:10.1016/j.gene.2004.11.028.Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 2013, 8, e55785, doi:10.1371/journal.pone.0055785.
- Gunter, T.E.; Gunter, K.K. Uptake of Calcium by Mitochondria: Transport and Possible Function. IUBMB Life (International Union Biochem. Mol. Biol. Life) 2001, 52, 197–204, doi:10.1080/15216540152846000.Kamer, K.J.; Grabarek, Z.; Mootha, V.K. High‐affinity cooperative Ca 2+ binding by MICU 1– MICU 2 serves as an on–off switch for the uniporter. EMBO Rep. 2017, 18, 1397–1411, doi:10.15252/embr.201643748.
- Xu, Z.; Zhang, D.; He, X.; Huang, Y.; Shao, H. Transport of Calcium Ions into Mitochondria. Curr. Genomics 2016, 17, 215–219, doi:10.2174/1389202917666160202215748.Payne, R.; Hoff, H.; Roskowski, A.; Foskett, J.K. MICU2 Restricts Spatial Crosstalk between InsP 3 R and MCU Channels by Regulating Threshold and Gain of MICU1-Mediated Inhibition and Activation of MCU. Cell Rep. 2017, 21, 3141–3154, doi:10.1016/j.celrep.2017.11.064.
- Sparagna, G.C.; Gunter, K.K.; Sheu, S.-S.; Gunter, T.E. Mitochondrial Calcium Uptake from Physiological-type Pulses of Calcium. J. Biol. Chem. 1995, 270, 27510–27515, doi:10.1074/jbc.270.46.27510.Mallilankaraman, K.; Cardenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenar, T.; Csordas, G.; Madireddi, P.; Yang, J.; Muller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343, doi:10.1038/ncb2622.
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340, doi:10.1038/nature10230.Ren, T.; Wang, J.; Zhang, H.; Yuan, P.; Zhu, J.; Wu, Y.; Huang, Q.; Guo, X.; Zhang, J.; Ji, L.; et al. MCUR1-Mediated Mitochondrial Calcium Signaling Facilitates Cell Survival of Hepatocellular Carcinoma via Reactive Oxygen Species-Dependent P53 Degradation. Antioxid. Redox Signal. 2018, 28, 1120–1136, doi:10.1089/ars.2017.6990.
- Beutner, G.; Sharma, V.K.; Giovannucci, D.R.; Yule, D.I.; Sheu, S.-S. Identification of a Ryanodine Receptor in Rat Heart Mitochondria. J. Biol. Chem. 2001, 276, 21482–21488, doi:10.1074/jbc.M101486200.Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S.J.; Breves, S.L.; Corbally, D.P.; et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016, 15, 1673–1685, doi:10.1016/j.celrep.2016.04.050.
- Beutner, G.; Sharma, V.K.; Lin, L.; Ryu, S.-Y.; Dirksen, R.T.; Sheu, S.-S. Type 1 ryanodine receptor in cardiac mitochondria: Transducer of excitation–metabolism coupling. Biochim. Biophys. Acta - Biomembr. 2005, 1717, 1–10, doi:10.1016/j.bbamem.2005.09.016.Paupe, V.; Prudent, J.; Dassa, E.P.; Rendon, O.Z.; Shoubridge, E.A. CCDC90A (MCUR1) Is a Cytochrome c Oxidase Assembly Factor and Not a Regulator of the Mitochondrial Calcium Uniporter. Cell Metab. 2015, 21, 109–116, doi:10.1016/j.cmet.2014.12.004.
- Babich, L.G.; Shlykov, S.G.; Kosterin, S.O. Ca ion transport in smooth muscle mitochondria. Ukr. Biochem. J. 2014, 86, 18–30, doi:10.15407/ubj86.06.018.Bassi, M.T.; Manzoni, M.; Bresciani, R.; Pizzo, M.T.; Della Monica, A.; Barlati, S.; Monti, E.; Borsani, G. Cellular expression and alternative splicing of SLC25A23, a member of the mitochondrial Ca2+-dependent solute carrier gene family. Gene 2005, 345, 173–182, doi:10.1016/j.gene.2004.11.028.
- Altschafl, B.A.; Beutner, G.; Sharma, V.K.; Sheu, S.-S.; Valdivia, H.H. The mitochondrial ryanodine receptor in rat heart: A pharmaco-kinetic profile. Biochim. Biophys. Acta - Biomembr. 2007, 1768, 1784–1795, doi:10.1016/j.bbamem.2007.04.011.Harborne, S.P.D.; King, M.S.; Crichton, P.G.; Kunji, E.R.S. Calcium regulation of the human mitochondrial ATP-Mg/Pi carrier SLC25A24 uses a locking pin mechanism. Sci. Rep. 2017, 7, 45383, doi:10.1038/srep45383.
- Tamai, S.; Iida, H.; Yokota, S.; Sayano, T.; Kiguchiya, S.; Ishihara, N.; Hayashi, J.-I.; Mihara, K.; Oka, T. Characterization of the mitochondrial protein LETM1, which maintains the mitochondrial tubular shapes and interacts with the AAA-ATPase BCS1L. J. Cell Sci. 2008, 121, 2588–2600, doi:10.1242/jcs.026625.Gunter, T.E.; Gunter, K.K. Uptake of Calcium by Mitochondria: Transport and Possible Function. IUBMB Life (International Union Biochem. Mol. Biol. Life) 2001, 52, 197–204, doi:10.1080/15216540152846000.
- Endele, S.; Fuhry, M.; Pak, S.J.; Zabel, B.U.; Winterpacht, A. LETM1, a novel gene encoding a putative EF-hand Ca(2+)-binding protein, flanks the Wolf-Hirschhorn syndrome (WHS) critical region and is deleted in most WHS patients. Genomics 1999, 60, 218–225, doi:10.1006/geno.1999.5881.Xu, Z.; Zhang, D.; He, X.; Huang, Y.; Shao, H. Transport of Calcium Ions into Mitochondria. Curr. Genomics 2016, 17, 215–219, doi:10.2174/1389202917666160202215748.
- Hasegawa, A.; van der Bliek, A.M. Inverse correlation between expression of the Wolfs Hirschhorn candidate gene Letm1 and mitochondrial volume in C. elegans and in mammalian cells. Hum. Mol. Genet. 2007, 16, 2061–2071, doi:10.1093/hmg/ddm154.Sparagna, G.C.; Gunter, K.K.; Sheu, S.-S.; Gunter, T.E. Mitochondrial Calcium Uptake from Physiological-type Pulses of Calcium. J. Biol. Chem. 1995, 270, 27510–27515, doi:10.1074/jbc.270.46.27510.
- Schlickum, S.; Moghekar, A.; Simpson, J.C.; Steglich, C.; O’Brien, R.J.; Winterpacht, A.; Endele, S.U. LETM1, a gene deleted in Wolf-Hirschhorn syndrome, encodes an evolutionarily conserved mitochondrial protein. Genomics 2004, 83, 254–261, doi:10.1016/j.ygeno.2003.08.013.Beutner, G.; Sharma, V.K.; Giovannucci, D.R.; Yule, D.I.; Sheu, S.-S. Identification of a Ryanodine Receptor in Rat Heart Mitochondria. J. Biol. Chem. 2001, 276, 21482–21488, doi:10.1074/jbc.M101486200.
- Lin, Q.-T.; Stathopulos, P.B. Molecular Mechanisms of Leucine Zipper EF-Hand Containing Transmembrane Protein-1 Function in Health and Disease. Int. J. Mol. Sci. 2019, 20, doi:10.3390/ijms20020286.Beutner, G.; Sharma, V.K.; Lin, L.; Ryu, S.-Y.; Dirksen, R.T.; Sheu, S.-S. Type 1 ryanodine receptor in cardiac mitochondria: Transducer of excitation–metabolism coupling. Biochim. Biophys. Acta - Biomembr. 2005, 1717, 1–10, doi:10.1016/j.bbamem.2005.09.016.
- Doonan, P.J.; Chandramoorthy, H.C.; Hoffman, N.E.; Zhang, X.; Cardenas, C.; Shanmughapriya, S.; Rajan, S.; Vallem, S.; Chen, X.; Foskett, J.K.; et al. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 4936–4949, doi:10.1096/fj.14-256453.Babich, L.G.; Shlykov, S.G.; Kosterin, S.O. Ca ion transport in smooth muscle mitochondria. Ukr. Biochem. J. 2014, 86, 18–30, doi:10.15407/ubj86.06.018.
- Jiang, D.; Zhao, L.; Clish, C.B.; Clapham, D.E. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E2249-54, doi:10.1073/pnas.1308558110.Altschafl, B.A.; Beutner, G.; Sharma, V.K.; Sheu, S.-S.; Valdivia, H.H. The mitochondrial ryanodine receptor in rat heart: A pharmaco-kinetic profile. Biochim. Biophys. Acta - Biomembr. 2007, 1768, 1784–1795, doi:10.1016/j.bbamem.2007.04.011.
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147, doi:10.1126/science.1175145.Tamai, S.; Iida, H.; Yokota, S.; Sayano, T.; Kiguchiya, S.; Ishihara, N.; Hayashi, J.-I.; Mihara, K.; Oka, T. Characterization of the mitochondrial protein LETM1, which maintains the mitochondrial tubular shapes and interacts with the AAA-ATPase BCS1L. J. Cell Sci. 2008, 121, 2588–2600, doi:10.1242/jcs.026625.
- Okamura, K.; Matsushita, S.; Kato, Y.; Watanabe, H.; Matsui, A.; Oka, T.; Matsuura, T. In vitro synthesis of the human calcium transporter Letm1 within cell-sized liposomes and investigation of its lipid dependency. J. Biosci. Bioeng. 2019, 127, 544–548, doi:10.1016/j.jbiosc.2018.11.003.Endele, S.; Fuhry, M.; Pak, S.J.; Zabel, B.U.; Winterpacht, A. LETM1, a novel gene encoding a putative EF-hand Ca(2+)-binding protein, flanks the Wolf-Hirschhorn syndrome (WHS) critical region and is deleted in most WHS patients. Genomics 1999, 60, 218–225, doi:10.1006/geno.1999.5881.
- Nowikovsky, K.; Bernardi, P. LETM1 in mitochondrial cation transport. Front. Physiol. 2014, 5, 83, doi:10.3389/fphys.2014.00083.Hasegawa, A.; van der Bliek, A.M. Inverse correlation between expression of the Wolfs Hirschhorn candidate gene Letm1 and mitochondrial volume in C. elegans and in mammalian cells. Hum. Mol. Genet. 2007, 16, 2061–2071, doi:10.1093/hmg/ddm154.
- De Marchi, U.; Santo-Domingo, J.; Castelbou, C.; Sekler, I.; Wiederkehr, A.; Demaurex, N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 2014, 289, 20377–20385, doi:10.1074/jbc.M113.540898.Schlickum, S.; Moghekar, A.; Simpson, J.C.; Steglich, C.; O’Brien, R.J.; Winterpacht, A.; Endele, S.U. LETM1, a gene deleted in Wolf-Hirschhorn syndrome, encodes an evolutionarily conserved mitochondrial protein. Genomics 2004, 83, 254–261, doi:10.1016/j.ygeno.2003.08.013.
- Carafoli, E.; Tiozzo, R.; Lugli, G.; Crovetti, F.; Kratzing, C. The release of calcium from heart mitochondria by sodium. J. Mol. Cell. Cardiol. 1974, 6, 361—371, doi:10.1016/0022-2828(74)90077-7.Lin, Q.-T.; Stathopulos, P.B. Molecular Mechanisms of Leucine Zipper EF-Hand Containing Transmembrane Protein-1 Function in Health and Disease. Int. J. Mol. Sci. 2019, 20, doi:10.3390/ijms20020286.
- Hunter, D.R.; Haworth, R.A.; Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria. Arch. Biochem. Biophys. 1979, 195, 468–477, doi:10.1016/0003-9861(79)90373-4.Doonan, P.J.; Chandramoorthy, H.C.; Hoffman, N.E.; Zhang, X.; Cardenas, C.; Shanmughapriya, S.; Rajan, S.; Vallem, S.; Chen, X.; Foskett, J.K.; et al. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 4936–4949, doi:10.1096/fj.14-256453.
- Nowikovsky, K.; Pozzan, T.; Rizzuto, R.; Scorrano, L.; Bernardi, P. The Pathophysiology of LETM1. J. Gen. Physiol. 2012, 139, 445–454, doi:10.1085/jgp.201110757.Jiang, D.; Zhao, L.; Clish, C.B.; Clapham, D.E. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E2249-54, doi:10.1073/pnas.1308558110.
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 436–441, doi:10.1073/pnas.0908099107.Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147, doi:10.1126/science.1175145.
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 2017, 545, 93–97, doi:10.1038/nature22082.Okamura, K.; Matsushita, S.; Kato, Y.; Watanabe, H.; Matsui, A.; Oka, T.; Matsuura, T. In vitro synthesis of the human calcium transporter Letm1 within cell-sized liposomes and investigation of its lipid dependency. J. Biosci. Bioeng. 2019, 127, 544–548, doi:10.1016/j.jbiosc.2018.11.003.
- Kostic, M.; Sekler, I. Functional properties and mode of regulation of the mitochondrial Na+/Ca2+ exchanger, NCLX. Semin. Cell Dev. Biol. 2019, 94, 59–65, doi:10.1016/j.semcdb.2019.01.009.Nowikovsky, K.; Bernardi, P. LETM1 in mitochondrial cation transport. Front. Physiol. 2014, 5, 83, doi:10.3389/fphys.2014.00083.
- Palty, R.; Ohana, E.; Hershfinkel, M.; Volokita, M.; Elgazar, V.; Beharier, O.; Silverman, W.F.; Argaman, M.; Sekler, I. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J. Biol. Chem. 2004, 279, 25234—25240, doi:10.1074/jbc.m401229200.De Marchi, U.; Santo-Domingo, J.; Castelbou, C.; Sekler, I.; Wiederkehr, A.; Demaurex, N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 2014, 289, 20377–20385, doi:10.1074/jbc.M113.540898.
- Sheng, J.-Z.; Prinsen, C.F.M.; Clark, R.B.; Giles, W.R.; Schnetkamp, P.P.M. Na+-Ca2+-K+ Currents Measured in Insect Cells Transfected with the Retinal Cone or Rod Na+-Ca2+-K+ Exchanger cDNA. Biophys. J. 2000, 79, 1945–1953, doi:10.1016/S0006-3495(00)76443-5.Carafoli, E.; Tiozzo, R.; Lugli, G.; Crovetti, F.; Kratzing, C. The release of calcium from heart mitochondria by sodium. J. Mol. Cell. Cardiol. 1974, 6, 361—371, doi:10.1016/0022-2828(74)90077-7.
- Gunter, T.E.; Yule, D.I.; Gunter, K.K.; Eliseev, R.A.; Salter, J.D. Calcium and mitochondria. FEBS Lett. 2004, 567, 96–102, doi:10.1016/j.febslet.2004.03.071.Wingrove, D.E.; Gunter, T.E. Kinetics of mitochondrial calcium transport. I. Characteristics of the sodium-independent calcium efflux mechanism of liver mitochondria. J. Biol. Chem. 1986, 261, 15159–15165.
- Takeuchi, A.; Kim, B.; Matsuoka, S. The destiny of Ca2+ released by mitochondria. J. Physiol. Sci. 2015, 65, 11–24, doi:10.1007/s12576-014-0326-7.Wingrove, D.E.; Gunter, T.E. Kinetics of mitochondrial calcium transport. II. A kinetic description of the sodium-dependent calcium efflux mechanism of liver mitochondria and inhibition by ruthenium red and by tetraphenylphosphonium. J. Biol. Chem. 1986, 261, 15166–15171.
- Haworth, R.A.; Hunter, D.R.; Berkoff, H.A. Na + releases Ca 2+ from liver, kidney and lung mitochondria. FEBS Lett. 1980, 110, 216–218, doi:10.1016/0014-5793(80)80076-7.Hunter, D.R.; Haworth, R.A.; Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria. Arch. Biochem. Biophys. 1979, 195, 468–477, doi:10.1016/0003-9861(79)90373-4.
- Zhang, Y.; Lipton, P. Cytosolic Ca2+ changes during in vitro ischemia in rat hippocampal slices: major roles for glutamate and Na+-dependent Ca2+ release from mitochondria. J. Neurosci. 1999, 19, 3307–3315.Tsai, M.-F.; Jiang, D.; Zhao, L.; Clapham, D.; Miller, C. Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J. Gen. Physiol. 2014, 143, 67–73, doi:10.1085/jgp.201311096.
- Islam, M.M.; Takeuchi, A.; Matsuoka, S. Membrane current evoked by mitochondrial Na+–Ca2+ exchange in mouse heart. J. Physiol. Sci. 2020, 70, 24, doi:10.1186/s12576-020-00752-3.Nowikovsky, K.; Pozzan, T.; Rizzuto, R.; Scorrano, L.; Bernardi, P. The Pathophysiology of LETM1. J. Gen. Physiol. 2012, 139, 445–454, doi:10.1085/jgp.201110757.
- Samanta, K.; Mirams, G.R.; Parekh, A.B. Sequential forward and reverse transport of the Na+ Ca2+ exchanger generates Ca2+ oscillations within mitochondria. Nat. Commun. 2018, 9, 156, doi:10.1038/s41467-017-02638-2.Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 436–441, doi:10.1073/pnas.0908099107.
- Kolomiets, O. V.; Danylovych, Y. V.; Danylovych, H. V.; Kosterin, S.O. Ca(2+)/H(+)-exchange in myometrium mitochondria. Ukr. Biochem. J. 2014, 86, 41–48, doi:10.15407/ubj86.03.041.Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 2017, 545, 93–97, doi:10.1038/nature22082.
- Kandaurova, N.V. Ca2+-induced Changes of Membrane Potential of Myometrium Mitochondria, 2011.Kostic, M.; Sekler, I. Functional properties and mode of regulation of the mitochondrial Na+/Ca2+ exchanger, NCLX. Semin. Cell Dev. Biol. 2019, 94, 59–65, doi:10.1016/j.semcdb.2019.01.009.
- Gunter, K.K.; Zuscik, M.J.; Gunter, T.E. The Na(+)-independent Ca2+ efflux mechanism of liver mitochondria is not a passive Ca2+/2H+ exchanger. J. Biol. Chem. 1991, 266, 21640–21648.Palty, R.; Ohana, E.; Hershfinkel, M.; Volokita, M.; Elgazar, V.; Beharier, O.; Silverman, W.F.; Argaman, M.; Sekler, I. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J. Biol. Chem. 2004, 279, 25234—25240, doi:10.1074/jbc.m401229200.
- Tsai, M.-F.; Jiang, D.; Zhao, L.; Clapham, D.; Miller, C. Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J. Gen. Physiol. 2014, 143, 67–73, doi:10.1085/jgp.201311096.Sheng, J.-Z.; Prinsen, C.F.M.; Clark, R.B.; Giles, W.R.; Schnetkamp, P.P.M. Na+-Ca2+-K+ Currents Measured in Insect Cells Transfected with the Retinal Cone or Rod Na+-Ca2+-K+ Exchanger cDNA. Biophys. J. 2000, 79, 1945–1953, doi:10.1016/S0006-3495(00)76443-5.
- Huang, E.; Qu, D.; Huang, T.; Rizzi, N.; Boonying, W.; Krolak, D.; Ciana, P.; Woulfe, J.; Klein, C.; Slack, R.S.; et al. PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat. Commun. 2017, 8, 1399, doi:10.1038/s41467-017-01435-1.Gunter, T.E.; Yule, D.I.; Gunter, K.K.; Eliseev, R.A.; Salter, J.D. Calcium and mitochondria. FEBS Lett. 2004, 567, 96–102, doi:10.1016/j.febslet.2004.03.071.
- Pérez, M.J.; Quintanilla, R.A. Development or disease: duality of the mitochondrial permeability transition pore. Dev. Biol. 2017, 426, 1–7, doi:10.1016/j.ydbio.2017.04.018.Takeuchi, A.; Kim, B.; Matsuoka, S. The destiny of Ca2+ released by mitochondria. J. Physiol. Sci. 2015, 65, 11–24, doi:10.1007/s12576-014-0326-7.
- Biasutto, L.; Azzolini, M.; Szabò, I.; Zoratti, M. The mitochondrial permeability transition pore in AD 2016: An update. Biochim. Biophys. Acta - Mol. Cell Res. 2016, 1863, 2515–2530, doi:10.1016/j.bbamcr.2016.02.012.Haworth, R.A.; Hunter, D.R.; Berkoff, H.A. Na + releases Ca 2+ from liver, kidney and lung mitochondria. FEBS Lett. 1980, 110, 216–218, doi:10.1016/0014-5793(80)80076-7.
- Li, Y.; Sun, J.; Wu, R.; Bai, J.; Hou, Y.; Zeng, Y.; Zhang, Y.; Wang, X.; Wang, Z.; Meng, X. Mitochondrial MPTP: A Novel Target of Ethnomedicine for Stroke Treatment by Apoptosis Inhibition. Front. Pharmacol. 2020, 11, 352, doi:10.3389/fphar.2020.00352.Zhang, Y.; Lipton, P. Cytosolic Ca2+ changes during in vitro ischemia in rat hippocampal slices: major roles for glutamate and Na+-dependent Ca2+ release from mitochondria. J. Neurosci. 1999, 19, 3307–3315.
- Hurst, S.; Hoek, J.; Sheu, S.-S. Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 27–47, doi:10.1007/s10863-016-9672-x.Islam, M.M.; Takeuchi, A.; Matsuoka, S. Membrane current evoked by mitochondrial Na+–Ca2+ exchange in mouse heart. J. Physiol. Sci. 2020, 70, 24, doi:10.1186/s12576-020-00752-3.
- Altschuld, R.A.; Hohl, C.M.; Castillo, L.C.; Garleb, A.A.; Starling, R.C.; Brierley, G.P. Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am. J. Physiol. Circ. Physiol. 1992, 262, H1699–H1704, doi:10.1152/ajpheart.1992.262.6.H1699.Samanta, K.; Mirams, G.R.; Parekh, A.B. Sequential forward and reverse transport of the Na+ Ca2+ exchanger generates Ca2+ oscillations within mitochondria. Nat. Commun. 2018, 9, 156, doi:10.1038/s41467-017-02638-2.
- Mnatsakanyan, N.; Beutner, G.; Porter, G.A.; Alavian, K.N.; Jonas, E.A. Physiological roles of the mitochondrial permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 13–25, doi:10.1007/s10863-016-9652-1.Kolomiets, O. V.; Danylovych, Y. V.; Danylovych, H. V.; Kosterin, S.O. Ca(2+)/H(+)-exchange in myometrium mitochondria. Ukr. Biochem. J. 2014, 86, 41–48, doi:10.15407/ubj86.03.041.
- Varanyuwatana, P.; Halestrap, A.P. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 2012, 12, 120–125, doi:10.1016/j.mito.2011.04.006.Kandaurova, N.V. Ca2+-induced Changes of Membrane Potential of Myometrium Mitochondria, 2011.
- Hurst, S.; Baggett, A.; Csordas, G.; Sheu, S.-S. SPG7 targets the m-AAA protease complex to process MCU for uniporter assembly, Ca 2+ influx, and regulation of mitochondrial permeability transition pore opening. J. Biol. Chem. 2019, 294, 10807–10818, doi:10.1074/jbc.RA118.006443.Gunter, K.K.; Zuscik, M.J.; Gunter, T.E. The Na(+)-independent Ca2+ efflux mechanism of liver mitochondria is not a passive Ca2+/2H+ exchanger. J. Biol. Chem. 1991, 266, 21640–21648.
- Leung, A.W.C.; Varanyuwatana, P.; Halestrap, A.P. The Mitochondrial Phosphate Carrier Interacts with Cyclophilin D and May Play a Key Role in the Permeability Transition. J. Biol. Chem. 2008, 283, 26312–26323, doi:10.1074/jbc.M805235200.Huang, E.; Qu, D.; Huang, T.; Rizzi, N.; Boonying, W.; Krolak, D.; Ciana, P.; Woulfe, J.; Klein, C.; Slack, R.S.; et al. PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat. Commun. 2017, 8, 1399, doi:10.1038/s41467-017-01435-1.
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabo, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. 2013, 110, 5887–5892, doi:10.1073/pnas.1217823110.Pérez, M.J.; Quintanilla, R.A. Development or disease: duality of the mitochondrial permeability transition pore. Dev. Biol. 2017, 426, 1–7, doi:10.1016/j.ydbio.2017.04.018.
- Bonora, M.; Bononi, A.; De Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the F O ATP synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683, doi:10.4161/cc.23599.Britti, E.; Delaspre, F.; Tamarit, J.; Ros, J. Mitochondrial calcium signalling and neurodegenerative diseases. Neuronal Signal. 2018, 2, doi:10.1042/NS20180061.
- CROMPTON, M.; COSTI, A. Kinetic evidence for a heart mitochondrial pore activated by Ca2+, inorganic phosphate and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular Ca2+ overload. Eur. J. Biochem. 1988, 178, 489–501, doi:10.1111/j.1432-1033.1988.tb14475.x.Biasutto, L.; Azzolini, M.; Szabò, I.; Zoratti, M. The mitochondrial permeability transition pore in AD 2016: An update. Biochim. Biophys. Acta - Mol. Cell Res. 2016, 1863, 2515–2530, doi:10.1016/j.bbamcr.2016.02.012.
- Carraro, M.; Giorgio, V.; Šileikytė, J.; Sartori, G.; Forte, M.; Lippe, G.; Zoratti, M.; Szabò, I.; Bernardi, P. Channel Formation by Yeast F-ATP Synthase and the Role of Dimerization in the Mitochondrial Permeability Transition. J. Biol. Chem. 2014, 289, 15980–15985, doi:10.1074/jbc.C114.559633.Li, Y.; Sun, J.; Wu, R.; Bai, J.; Hou, Y.; Zeng, Y.; Zhang, Y.; Wang, X.; Wang, Z.; Meng, X. Mitochondrial MPTP: A Novel Target of Ethnomedicine for Stroke Treatment by Apoptosis Inhibition. Front. Pharmacol. 2020, 11, 352, doi:10.3389/fphar.2020.00352.
- Zhou, W.; Marinelli, F.; Nief, C.; Faraldo-Gómez, J.D. Atomistic simulations indicate the c-subunit ring of the F1Fo ATP synthase is not the mitochondrial permeability transition pore. Elife 2017, 6, doi:10.7554/eLife.23781.Hurst, S.; Hoek, J.; Sheu, S.-S. Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 27–47, doi:10.1007/s10863-016-9672-x.
- He, J.; Ford, H.C.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc. Natl. Acad. Sci. 2017, 114, 3409–3414, doi:10.1073/pnas.1702357114.Altschuld, R.A.; Hohl, C.M.; Castillo, L.C.; Garleb, A.A.; Starling, R.C.; Brierley, G.P. Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am. J. Physiol. Circ. Physiol. 1992, 262, H1699–H1704, doi:10.1152/ajpheart.1992.262.6.H1699.
- He, J.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc. Natl. Acad. Sci. 2017, 114, 9086–9091, doi:10.1073/pnas.1711201114.Mnatsakanyan, N.; Beutner, G.; Porter, G.A.; Alavian, K.N.; Jonas, E.A. Physiological roles of the mitochondrial permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 13–25, doi:10.1007/s10863-016-9652-1.
- Bonora, M.; Wieckowski, M.R.; Chinopoulos, C.; Kepp, O.; Kroemer, G.; Galluzzi, L.; Pinton, P. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 2015, 34, 1475–1486, doi:10.1038/onc.2014.96.Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the Permeability Transition Pore in Mitochondria Devoid of Cyclophilin D. J. Biol. Chem. 2005, 280, 18558–18561, doi:10.1074/jbc.C500089200.
- Halestrap, A.P.; Richardson, A.P. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 129–141, doi:10.1016/j.yjmcc.2014.08.018.Shanmughapriya, S.; Rajan, S.; Hoffman, N.E.; Higgins, A.M.; Tomar, D.; Nemani, N.; Hines, K.J.; Smith, D.J.; Eguchi, A.; Vallem, S.; et al. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol. Cell 2015, 60, 47–62, doi:10.1016/j.molcel.2015.08.009.
- Bernardi, P. The mitochondrial permeability transition pore: a mystery solved? Front. Physiol. 2013, 4, doi:10.3389/fphys.2013.00095.Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662, doi:10.1038/nature03434.
- Jonas, E.A.; Porter, G.A.; Beutner, G.; Mnatsakanyan, N.; Alavian, K.N. Cell death disguised: The mitochondrial permeability transition pore as the c-subunit of the F1FO ATP synthase. Pharmacol. Res. 2015, 99, 382–392, doi:10.1016/j.phrs.2015.04.013.Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658, doi:10.1038/nature03317.
- Elustondo, P.A.; Nichols, M.; Negoda, A.; Thirumaran, A.; Zakharian, E.; Robertson, G.S.; Pavlov, E. V Mitochondrial permeability transition pore induction is linked to formation of the complex of ATPase C-subunit, polyhydroxybutyrate and inorganic polyphosphate. Cell Death Discov. 2016, 2, 16070, doi:10.1038/cddiscovery.2016.70.Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. 2005, 102, 12005–12010, doi:10.1073/pnas.0505294102.
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.-A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. 2014, 111, 10580–10585, doi:10.1073/pnas.1401591111.Alam, M.R.; Baetz, D.; Ovize, M. Cyclophilin D and myocardial ischemia–reperfusion injury: A fresh perspective. J. Mol. Cell. Cardiol. 2015, 78, 80–89, doi:10.1016/j.yjmcc.2014.09.026.
- Chinopoulos, C. Mitochondrial permeability transition pore: Back to the drawing board. Neurochem. Int. 2018, 117, 49–54, doi:10.1016/j.neuint.2017.06.010.Kokoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465, doi:10.1038/nature02229.
- Ichas, F.; Jouaville, L.S.; Mazat, J.-P. Mitochondria Are Excitable Organelles Capable of Generating and Conveying Electrical and Calcium Signals. Cell 1997, 89, 1145–1153, doi:10.1016/S0092-8674(00)80301-3.Karch, J.; Bround, M.J.; Khalil, H.; Sargent, M.A.; Latchman, N.; Terada, N.; Peixoto, P.M.; Molkentin, J.D. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 2019, 5, eaaw4597, doi:10.1126/sciadv.aaw4597.
- Lu, X.; Kwong, J.Q.; Molkentin, J.D.; Bers, D.M. Individual Cardiac Mitochondria Undergo Rare Transient Permeability Transition Pore Openings. Circ. Res. 2016, 118, 834–841, doi:10.1161/CIRCRESAHA.115.308093.Bround, M.J.; Bers, D.M.; Molkentin, J.D. A 20/20 view of ANT function in mitochondrial biology and necrotic cell death. J. Mol. Cell. Cardiol. 2020, 144, A3–A13, doi:10.1016/j.yjmcc.2020.05.012.
- Ichas, F.; Mazat, J.-P. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta - Bioenerg. 1998, 1366, 33–50, doi:10.1016/S0005-2728(98)00119-4.Kwong, J.Q.; Davis, J.; Baines, C.P.; Sargent, M.A.; Karch, J.; Wang, X.; Huang, T.; Molkentin, J.D. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014, 21, 1209–1217, doi:10.1038/cdd.2014.36.
- Gainutdinov, T.; Molkentin, J.D.; Siemen, D.; Ziemer, M.; Debska-Vielhaber, G.; Vielhaber, S.; Gizatullina, Z.; Orynbayeva, Z.; Gellerich, F.N. Knockout of cyclophilin D in Ppif−/− mice increases stability of brain mitochondria against Ca2+ stress. Arch. Biochem. Biophys. 2015, 579, 40–46, doi:10.1016/j.abb.2015.05.009.Varanyuwatana, P.; Halestrap, A.P. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 2012, 12, 120–125, doi:10.1016/j.mito.2011.04.006.
- Bernardi, P.; von Stockum, S. The permeability transition pore as a Ca2+ release channel: New answers to an old question. Cell Calcium 2012, 52, 22–27, doi:10.1016/j.ceca.2012.03.004.Hurst, S.; Baggett, A.; Csordas, G.; Sheu, S.-S. SPG7 targets the m-AAA protease complex to process MCU for uniporter assembly, Ca 2+ influx, and regulation of mitochondrial permeability transition pore opening. J. Biol. Chem. 2019, 294, 10807–10818, doi:10.1074/jbc.RA118.006443.
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the Permeability Transition Pore in Mitochondria Devoid of Cyclophilin D. J. Biol. Chem. 2005, 280, 18558–18561, doi:10.1074/jbc.C500089200.Leung, A.W.C.; Varanyuwatana, P.; Halestrap, A.P. The Mitochondrial Phosphate Carrier Interacts with Cyclophilin D and May Play a Key Role in the Permeability Transition. J. Biol. Chem. 2008, 283, 26312–26323, doi:10.1074/jbc.M805235200.
- Shanmughapriya, S.; Rajan, S.; Hoffman, N.E.; Higgins, A.M.; Tomar, D.; Nemani, N.; Hines, K.J.; Smith, D.J.; Eguchi, A.; Vallem, S.; et al. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol. Cell 2015, 60, 47–62, doi:10.1016/j.molcel.2015.08.009.Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabo, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. 2013, 110, 5887–5892, doi:10.1073/pnas.1217823110.
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662, doi:10.1038/nature03434.Bonora, M.; Bononi, A.; De Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the F O ATP synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683, doi:10.4161/cc.23599.
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658, doi:10.1038/nature03317.CROMPTON, M.; COSTI, A. Kinetic evidence for a heart mitochondrial pore activated by Ca2+, inorganic phosphate and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular Ca2+ overload. Eur. J. Biochem. 1988, 178, 489–501, doi:10.1111/j.1432-1033.1988.tb14475.x.
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. 2005, 102, 12005–12010, doi:10.1073/pnas.0505294102.Carraro, M.; Giorgio, V.; Šileikytė, J.; Sartori, G.; Forte, M.; Lippe, G.; Zoratti, M.; Szabò, I.; Bernardi, P. Channel Formation by Yeast F-ATP Synthase and the Role of Dimerization in the Mitochondrial Permeability Transition. J. Biol. Chem. 2014, 289, 15980–15985, doi:10.1074/jbc.C114.559633.
- Alam, M.R.; Baetz, D.; Ovize, M. Cyclophilin D and myocardial ischemia–reperfusion injury: A fresh perspective. J. Mol. Cell. Cardiol. 2015, 78, 80–89, doi:10.1016/j.yjmcc.2014.09.026.Zhou, W.; Marinelli, F.; Nief, C.; Faraldo-Gómez, J.D. Atomistic simulations indicate the c-subunit ring of the F1Fo ATP synthase is not the mitochondrial permeability transition pore. Elife 2017, 6, doi:10.7554/eLife.23781.
- Kokoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465, doi:10.1038/nature02229.He, J.; Ford, H.C.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc. Natl. Acad. Sci. 2017, 114, 3409–3414, doi:10.1073/pnas.1702357114.
- Karch, J.; Bround, M.J.; Khalil, H.; Sargent, M.A.; Latchman, N.; Terada, N.; Peixoto, P.M.; Molkentin, J.D. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 2019, 5, eaaw4597, doi:10.1126/sciadv.aaw4597.He, J.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc. Natl. Acad. Sci. 2017, 114, 9086–9091, doi:10.1073/pnas.1711201114.
- Bround, M.J.; Bers, D.M.; Molkentin, J.D. A 20/20 view of ANT function in mitochondrial biology and necrotic cell death. J. Mol. Cell. Cardiol. 2020, 144, A3–A13, doi:10.1016/j.yjmcc.2020.05.012.Bonora, M.; Wieckowski, M.R.; Chinopoulos, C.; Kepp, O.; Kroemer, G.; Galluzzi, L.; Pinton, P. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 2015, 34, 1475–1486, doi:10.1038/onc.2014.96.
- Kwong, J.Q.; Davis, J.; Baines, C.P.; Sargent, M.A.; Karch, J.; Wang, X.; Huang, T.; Molkentin, J.D. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014, 21, 1209–1217, doi:10.1038/cdd.2014.36.Halestrap, A.P.; Richardson, A.P. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 129–141, doi:10.1016/j.yjmcc.2014.08.018.
- Korge, P.; Yang, L.; Yang, J.-H.; Wang, Y.; Qu, Z.; Weiss, J.N. Protective Role of Transient Pore Openings in Calcium Handling by Cardiac Mitochondria. J. Biol. Chem. 2011, 286, 34851–34857, doi:10.1074/jbc.M111.239921.Bernardi, P. The mitochondrial permeability transition pore: a mystery solved? Front. Physiol. 2013, 4, doi:10.3389/fphys.2013.00095.
- Elrod, J.W.; Wong, R.; Mishra, S.; Vagnozzi, R.J.; Sakthievel, B.; Goonasekera, S.A.; Karch, J.; Gabel, S.; Farber, J.; Force, T.; et al. Cyclophilin D controls mitochondrial pore–dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 2010, 120, 3680–3687, doi:10.1172/JCI43171.Jonas, E.A.; Porter, G.A.; Beutner, G.; Mnatsakanyan, N.; Alavian, K.N. Cell death disguised: The mitochondrial permeability transition pore as the c-subunit of the F1FO ATP synthase. Pharmacol. Res. 2015, 99, 382–392, doi:10.1016/j.phrs.2015.04.013.
- Lamb, H.M. Double agents of cell death: novel emerging functions of apoptotic regulators. FEBS J. 2020, doi:10.1111/febs.15308.Elustondo, P.A.; Nichols, M.; Negoda, A.; Thirumaran, A.; Zakharian, E.; Robertson, G.S.; Pavlov, E. V Mitochondrial permeability transition pore induction is linked to formation of the complex of ATPase C-subunit, polyhydroxybutyrate and inorganic polyphosphate. Cell Death Discov. 2016, 2, 16070, doi:10.1038/cddiscovery.2016.70.
- Baines, C.P.; Gutiérrez-Aguilar, M. The still uncertain identity of the channel-forming unit(s) of the mitochondrial permeability transition pore. Cell Calcium 2018, 73, 121–130, doi:10.1016/j.ceca.2018.05.003.Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.-A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. 2014, 111, 10580–10585, doi:10.1073/pnas.1401591111.
- Zorow, D.B.; Kinnally, K.W.; Perini, S.; Tedeschi, H. Multiple conductance levels in rat heart inner mitochondrial membranes studied by patch clamping. Biochim. Biophys. Acta - Biomembr. 1992, 1105, 263–270, doi:10.1016/0005-2736(92)90203-X.Chinopoulos, C. Mitochondrial permeability transition pore: Back to the drawing board. Neurochem. Int. 2018, 117, 49–54, doi:10.1016/j.neuint.2017.06.010.
- Petronilli, V.; Miotto, G.; Canton, M.; Brini, M.; Colonna, R.; Bernardi, P.; Di Lisa, F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 1999, 76, 725–734, doi:10.1016/S0006-3495(99)77239-5.Ichas, F.; Jouaville, L.S.; Mazat, J.-P. Mitochondria Are Excitable Organelles Capable of Generating and Conveying Electrical and Calcium Signals. Cell 1997, 89, 1145–1153, doi:10.1016/S0092-8674(00)80301-3.
- Elrod, J.W.; Molkentin, J.D. Physiologic Functions of Cyclophilin D and the Mitochondrial Permeability Transition Pore. Circ. J. 2013, 77, 1111–1122, doi:10.1253/circj.CJ-13-0321.Lu, X.; Kwong, J.Q.; Molkentin, J.D.; Bers, D.M. Individual Cardiac Mitochondria Undergo Rare Transient Permeability Transition Pore Openings. Circ. Res. 2016, 118, 834–841, doi:10.1161/CIRCRESAHA.115.308093.
- Halestrap, A. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385, doi:10.1016/S0008-6363(03)00533-9.Ichas, F.; Mazat, J.-P. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta - Bioenerg. 1998, 1366, 33–50, doi:10.1016/S0005-2728(98)00119-4.
- Szabó, I.; Zoratti, M. The mitochondrial permeability transition pore may comprise VDAC molecules. FEBS Lett. 1993, 330, 201–205, doi:10.1016/0014-5793(93)80273-W.Gainutdinov, T.; Molkentin, J.D.; Siemen, D.; Ziemer, M.; Debska-Vielhaber, G.; Vielhaber, S.; Gizatullina, Z.; Orynbayeva, Z.; Gellerich, F.N. Knockout of cyclophilin D in Ppif−/− mice increases stability of brain mitochondria against Ca2+ stress. Arch. Biochem. Biophys. 2015, 579, 40–46, doi:10.1016/j.abb.2015.05.009.
- Crompton, M.; Virji, S.; Ward, J.M. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 1998, 258, 729–735, doi:10.1046/j.1432-1327.1998.2580729.x.Bernardi, P.; von Stockum, S. The permeability transition pore as a Ca2+ release channel: New answers to an old question. Cell Calcium 2012, 52, 22–27, doi:10.1016/j.ceca.2012.03.004.
- Zheng, Y.; Shi, Y.; Tian, C.; Jiang, C.; Jin, H.; Chen, J.; Almasan, A.; Tang, H.; Chen, Q. Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene 2004, 23, 1239–1247, doi:10.1038/sj.onc.1207205.Korge, P.; Yang, L.; Yang, J.-H.; Wang, Y.; Qu, Z.; Weiss, J.N. Protective Role of Transient Pore Openings in Calcium Handling by Cardiac Mitochondria. J. Biol. Chem. 2011, 286, 34851–34857, doi:10.1074/jbc.M111.239921.
- Chaudhuri, A.D.; Choi, D.C.; Kabaria, S.; Tran, A.; Junn, E. MicroRNA-7 Regulates the Function of Mitochondrial Permeability Transition Pore by Targeting VDAC1 Expression. J. Biol. Chem. 2016, 291, 6483–6493, doi:10.1074/jbc.M115.691352.Elrod, J.W.; Wong, R.; Mishra, S.; Vagnozzi, R.J.; Sakthievel, B.; Goonasekera, S.A.; Karch, J.; Gabel, S.; Farber, J.; Force, T.; et al. Cyclophilin D controls mitochondrial pore–dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 2010, 120, 3680–3687, doi:10.1172/JCI43171.
- Zhou, H.; Hu, S.; Jin, Q.; Shi, C.; Zhang, Y.; Zhu, P.; Ma, Q.; Tian, F.; Chen, Y. Mff‐Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of mROS‐Mediated Cardiolipin Oxidation and HK2/VDAC1 Disassociation‐Involved mPTP Opening. J. Am. Heart Assoc. 2017, 6, doi:10.1161/JAHA.116.005328.Lamb, H.M. Double agents of cell death: novel emerging functions of apoptotic regulators. FEBS J. 2020, doi:10.1111/febs.15308.
- Tan, W.; Colombini, M. VDAC closure increases calcium ion flux. Biochim. Biophys. Acta - Biomembr. 2007, 1768, 2510–2515, doi:10.1016/j.bbamem.2007.06.002.Baines, C.P.; Gutiérrez-Aguilar, M. The still uncertain identity of the channel-forming unit(s) of the mitochondrial permeability transition pore. Cell Calcium 2018, 73, 121–130, doi:10.1016/j.ceca.2018.05.003.
- Tikunov, A.; Johnson, C.B.; Pediaditakis, P.; Markevich, N.; Macdonald, J.M.; Lemasters, J.J.; Holmuhamedov, E. Closure of VDAC causes oxidative stress and accelerates the Ca2+-induced mitochondrial permeability transition in rat liver mitochondria. Arch. Biochem. Biophys. 2010, 495, 174–181, doi:10.1016/j.abb.2010.01.008.Zorow, D.B.; Kinnally, K.W.; Perini, S.; Tedeschi, H. Multiple conductance levels in rat heart inner mitochondrial membranes studied by patch clamping. Biochim. Biophys. Acta - Biomembr. 1992, 1105, 263–270, doi:10.1016/0005-2736(92)90203-X.
- Xu, H.; Cui, S.; Zhang, Y.; Ren, J. Mitochondrial Ca2+ regulation in the etiology of heart failure: physiological and pathophysiological implications. Acta Pharmacol. Sin. 2020, doi:10.1038/s41401-020-0476-5.Petronilli, V.; Miotto, G.; Canton, M.; Brini, M.; Colonna, R.; Bernardi, P.; Di Lisa, F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 1999, 76, 725–734, doi:10.1016/S0006-3495(99)77239-5.
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.-Y.; Dejean, L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta - Mol. Cell Res. 2011, 1813, 616–622, doi:10.1016/j.bbamcr.2010.09.013.Xu, H.; Cui, S.; Zhang, Y.; Ren, J. Mitochondrial Ca2+ regulation in the etiology of heart failure: physiological and pathophysiological implications. Acta Pharmacol. Sin. 2020, doi:10.1038/s41401-020-0476-5.
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155, doi:10.1152/physrev.00001.2015.Kinnally, K.W.; Peixoto, P.M.; Ryu, S.-Y.; Dejean, L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta - Mol. Cell Res. 2011, 1813, 616–622, doi:10.1016/j.bbamcr.2010.09.013.
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155, doi:10.1152/physrev.00001.2015.
- Elrod, J.W.; Molkentin, J.D. Physiologic Functions of Cyclophilin D and the Mitochondrial Permeability Transition Pore. Circ. J. 2013, 77, 1111–1122, doi:10.1253/circj.CJ-13-0321.
- Halestrap, A. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385, doi:10.1016/S0008-6363(03)00533-9.
- Szabó, I.; Zoratti, M. The mitochondrial permeability transition pore may comprise VDAC molecules. FEBS Lett. 1993, 330, 201–205, doi:10.1016/0014-5793(93)80273-W.
- Crompton, M.; Virji, S.; Ward, J.M. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 1998, 258, 729–735, doi:10.1046/j.1432-1327.1998.2580729.x.
- Zheng, Y.; Shi, Y.; Tian, C.; Jiang, C.; Jin, H.; Chen, J.; Almasan, A.; Tang, H.; Chen, Q. Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene 2004, 23, 1239–1247, doi:10.1038/sj.onc.1207205.
- Chaudhuri, A.D.; Choi, D.C.; Kabaria, S.; Tran, A.; Junn, E. MicroRNA-7 Regulates the Function of Mitochondrial Permeability Transition Pore by Targeting VDAC1 Expression. J. Biol. Chem. 2016, 291, 6483–6493, doi:10.1074/jbc.M115.691352.
- Zhou, H.; Hu, S.; Jin, Q.; Shi, C.; Zhang, Y.; Zhu, P.; Ma, Q.; Tian, F.; Chen, Y. Mff‐Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of mROS‐Mediated Cardiolipin Oxidation and HK2/VDAC1 Disassociation‐Involved mPTP Opening. J. Am. Heart Assoc. 2017, 6, doi:10.1161/JAHA.116.005328.
- Tan, W.; Colombini, M. VDAC closure increases calcium ion flux. Biochim. Biophys. Acta - Biomembr. 2007, 1768, 2510–2515, doi:10.1016/j.bbamem.2007.06.002.
- Tikunov, A.; Johnson, C.B.; Pediaditakis, P.; Markevich, N.; Macdonald, J.M.; Lemasters, J.J.; Holmuhamedov, E. Closure of VDAC causes oxidative stress and accelerates the Ca2+-induced mitochondrial permeability transition in rat liver mitochondria. Arch. Biochem. Biophys. 2010, 495, 174–181, doi:10.1016/j.abb.2010.01.008.