Due to its intrinsic ability to expedite genetic gains achieved through improved selection accuracy coupled with reduced breeding time and phenotyping cost, GS has garnered huge attention over the past decade in wheat [40]. Various efforts have been undertaken to practically implement the GS strategy for abiotic stress tolerance, particularly heat and drought stress, and different robust statistical models have been tested to analyze their impact on prediction accuracies [41][42][43][44][45][46][47][48]. Additionally, attempts have been made to optimize genomic prediction accuracies for resistance against a wide range of pathogens in wheat, including powdery mildew (causative agent: Due to its intrinsic ability to expedite genetic gains achieved through improved selection accuracy coupled with reduced breeding time and phenotyping cost, GS has garnered huge attention over the past decade in wheat [85]. Various efforts have been undertaken to practically implement the GS strategy for abiotic stress tolerance, particularly heat and drought stress, and different robust statistical models have been tested to analyze their impact on prediction accuracies [77,90,91,92,93,94,95,96]. Additionally, attempts have been made to optimize genomic prediction accuracies for resistance against a wide range of pathogens in wheat, including powdery mildew (causative agent: Blumeria graminis; [49]), fusarium head blight (causative agent: ; [97]), fusarium head blight (causative agent: Fusarium graminearum; [50][51][52][53]), septoria tritici blotch (causative agent: ; [98,99,100,101]), septoria tritici blotch (causative agent: Zymoseptoriatritici; [51][54][55]), stem or black rust (causative agent: ; [99,102,103]), stem or black rust (causative agent: Puccinia graminis Pers; [56]), leaf or brown rust (causative agent: Pers; [104]), leaf or brown rust (causative agent: Puccinia triticina Eriks; [56]), stripe or yellow rust (causative agent: Eriks; [104]), stripe or yellow rust (causative agent: Puccinia striiformis West; [56][57]), stagonospora nodorum blotch (causative agent: West; [104,105]), stagonospora nodorum blotch (causative agent: Parastagonospora nodorum; [54]), spot blotch (causative agent: ; [102]), spot blotch (causative agent: Bipolaris sorokiniana; [58] and tan spot (causative agent: ; [32] and tan spot (causative agent: Pyrenophora tritici-repentis; [54]). Integrating omics datasets in prediction models and compounding GS with high-throughput phenotyping, speed breeding and gene editing techniques would be instrumental in hastening wheat improvement programs for the development of future climate-resilient, high-yielding superior wheat cultivars. ; [102]). Integrating omics datasets in prediction models and compounding GS with high-throughput phenotyping, speed breeding and gene editing techniques would be instrumental in hastening wheat improvement programs for the development of future climate-resilient, high-yielding superior wheat cultivars.
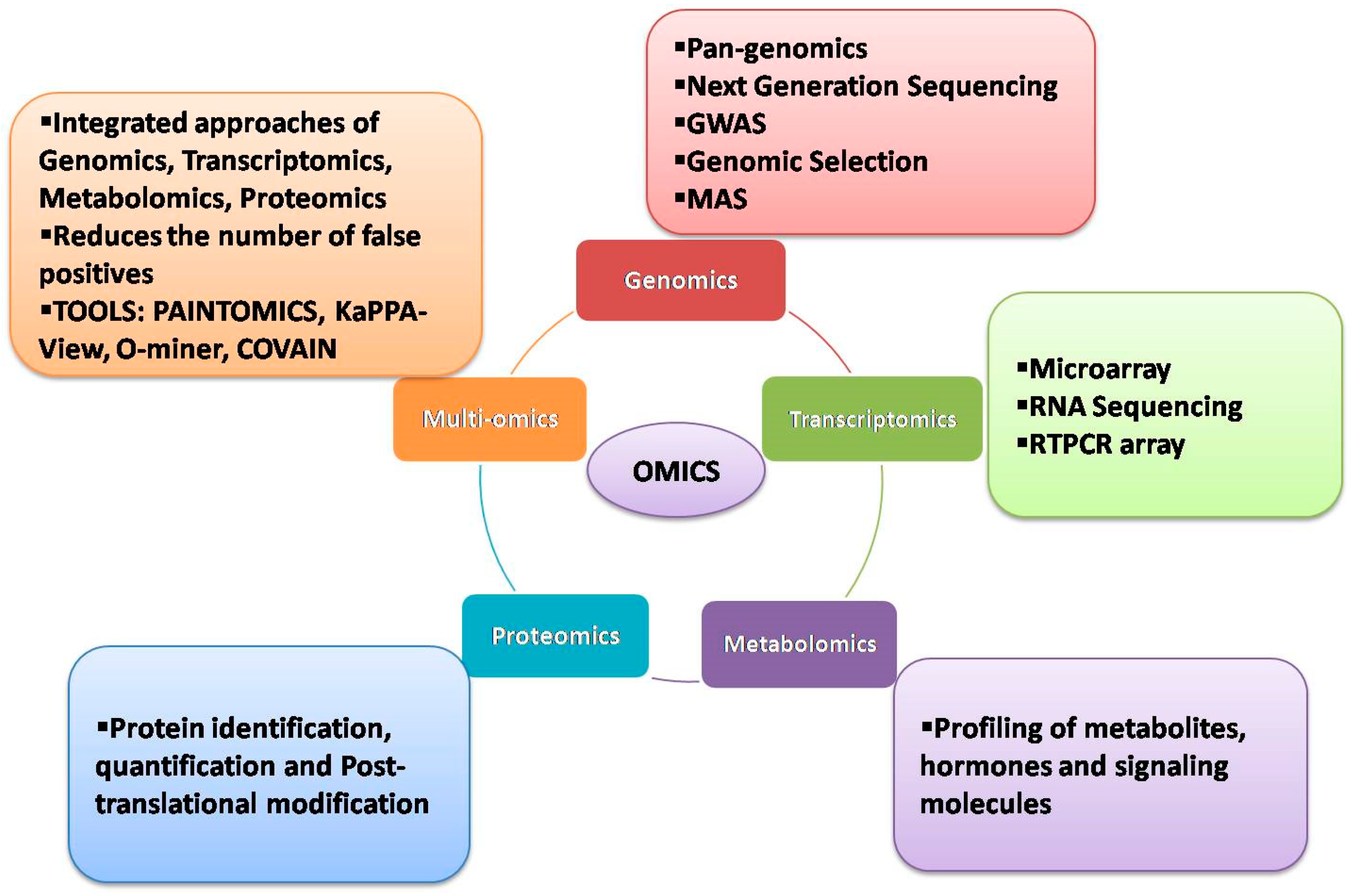
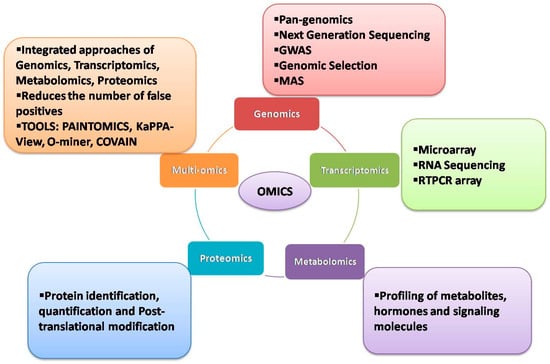
3. Transcriptomic Approaches
In the past, microarrays have been used extensively to analyze the expression and co-expression of numerous genes under various stress conditions in crop plants [59][60][61]. However, microarray-based experiments failed to detect gene networks regulating stress responses at the genome-wide level. With advancements in NGS, whole transcriptome analyses have become feasible, allowing identification and quantification of the global expression of transcripts, alternative splicing patterns and associated allele-specific expressions [62][63]. RNA-seq, the latest NGS technique for investigating genome-wide transcriptomes, helps to examine the expressional variation in genes in contrasting sets of samples or panels subjected to different stress treatments and to pinpoint the potential candidate genes. Due to its in-depth coverage and global expression of transcripts, RNA-seq has been used extensively in many crops, including wheat, to uncover the mechanisms conferring tolerance to different stresses.
In the past, microarrays have been used extensively to analyze the expression and co-expression of numerous genes under various stress conditions in crop plants [106,107,108]. However, microarray-based experiments failed to detect gene networks regulating stress responses at the genome-wide level. With advancements in NGS, whole transcriptome analyses have become feasible, allowing identification and quantification of the global expression of transcripts, alternative splicing patterns and associated allele-specific expressions [109,110]. RNA-seq, the latest NGS technique for investigating genome-wide transcriptomes, helps to examine the expressional variation in genes in contrasting sets of samples or panels subjected to different stress treatments and to pinpoint the potential candidate genes. Due to its in-depth coverage and global expression of transcripts, RNA-seq has been used extensively in many crops, including wheat, to uncover the mechanisms conferring tolerance to different stresses.
Transcriptomics studies have unveiled important roles played by numerous genes, gene families, transcription factors (TFs), hormones, metabolites, cofactors and microRNAs (miRNAs) in conferring tolerance to various abiotic stresses in wheat. Most significantly, the role of antioxidant enzymes, such as cytochrome P450, glutathione S-transferase, polyphenol oxidase, chitinase 2, ascorbate peroxidase (APX) and peroxidase (POD), in circumventing the adverse effects of reactive oxygen species (ROS) on proteins has been repeatedly suggested to be an important tolerance mechanism under both drought and salinity stress conditions [64][65][66]. Similarly, ABC transporters and Na Transcriptomics studies have unveiled important roles played by numerous genes, gene families, transcription factors (TFs), hormones, metabolites, cofactors and microRNAs (miRNAs) in conferring tolerance to various abiotic stresses in wheat. Most significantly, the role of antioxidant enzymes, such as cytochrome P450, glutathione S-transferase, polyphenol oxidase, chitinase 2, ascorbate peroxidase (APX) and peroxidase (POD), in circumventing the adverse effects of reactive oxygen species (ROS) on proteins has been repeatedly suggested to be an important tolerance mechanism under both drought and salinity stress conditions [122,123,124]. Similarly, ABC transporters and Na+
/Ca2+ exchangers have been suggested to be the key players under salinity stress conditions. In addition, the role of various TFs belonging to the NAC, WRKY and MADS families was highlighted to be a significant one under a single or multiple abiotic stress condition while TFs from the MYB family were highlighted as the key candidate genes under biotic stress conditions [67][68]. exchangers have been suggested to be the key players under salinity stress conditions. In addition, the role of various TFs belonging to the NAC, WRKY and MADS families was highlighted to be a significant one under a single or multiple abiotic stress condition while TFs from the MYB family were highlighted as the key candidate genes under biotic stress conditions [125,126].
The transcriptomic alterations in response to salt stress were analysed to identify the candidate genes regulating plant salt stress tolerance in wheat [67]. Two sets of genotypes, contrasting for sensitivity to salinity stress wherein one set consisted of a German winter wheat cultivar Zentos (salt-tolerant) and the synthetic genotype Syn86 (salt-susceptible), while the other set had a Turkish cultivar Altay2000 (salt-tolerant) and the Uzbek cultivar Bobur (salt-susceptible) were analysed [67]. The expression of genes from four genotypes revealed upregulation of some ABC transporters and Na The transcriptomic alterations in response to salt stress were analysed to identify the candidate genes regulating plant salt stress tolerance in wheat [125]. Two sets of genotypes, contrasting for sensitivity to salinity stress wherein one set consisted of a German winter wheat cultivar Zentos (salt-tolerant) and the synthetic genotype Syn86 (salt-susceptible), while the other set had a Turkish cultivar Altay2000 (salt-tolerant) and the Uzbek cultivar Bobur (salt-susceptible) were analysed [125]. The expression of genes from four genotypes revealed upregulation of some ABC transporters and Na+
/Ca2+ exchangers in the tolerant genotypes, indicating their involvement in the mechanisms of sodium exclusion and homeostasis. Furthermore, five genes were found co-located with two QTLs on chromosome 2A and three of these were found to have a differential expression. Of these, TraesCS2A02G395000, which codes for an oxoglutarate/iron-dependent dioxygenase, was reported to be central in controlling salinity stress in wheat [67]. Transcriptomic analyses in wheat was employed to assess regulation of biosynthesis of a special metabolite, benzoxazinoid, by the wheat transcription factor exchangers in the tolerant genotypes, indicating their involvement in the mechanisms of sodium exclusion and homeostasis. Furthermore, five genes were found co-located with two QTLs on chromosome 2A and three of these were found to have a differential expression. Of these, TraesCS2A02G395000, which codes for an oxoglutarate/iron-dependent dioxygenase, was reported to be central in controlling salinity stress in wheat [125]. Transcriptomic analyses in wheat was employed to assess regulation of biosynthesis of a special metabolite, benzoxazinoid, by the wheat transcription factor MYB31 in response to pest attack [68]. The silencing of in response to pest attack [126]. The silencing of TaMYB31 gene was found to significantly decrease the benzoxazinoid metabolite levels and consequently resulting in susceptibility to herbivores [68]. Furthermore, comprehensive transcriptomics highlighted the fact that the gene was found to significantly decrease the benzoxazinoid metabolite levels and consequently resulting in susceptibility to herbivores [126]. Furthermore, comprehensive transcriptomics highlighted the fact that the TaMYB31
gene co-expresses with the target benzoxazinoid-encoded Bx genes under various biotic and environmental circumstances imparting resistance to wheat plants [68]. RNAseq analysis of wheat lines subjected to multiple abiotic stresses revealed a novel ERF gene TaERF-6-3A, which was found to be induced under three abiotic stresses, salt, cold and drought [69]. In addition, upregulation of 20 genes under various biotic and environmental circumstances imparting resistance to wheat plants [126]. RNAseq analysis of wheat lines subjected to multiple abiotic stresses revealed a novel ERF gene TaERF-6-3A, which was found to be induced under three abiotic stresses, salt, cold and drought [127]. In addition, upregulation of 20 AP2/ERF genes was identified based in response to drought stress [69]. Most significantly, a novel genes was identified based in response to drought stress [127]. Most significantly, a novel ERF
gene, TaERF-6-3A, was found to be induced in response to three abiotic stresses, salt, cold and drought [69]. , was found to be induced in response to three abiotic stresses, salt, cold and drought [127].
Heavy metal contamination of the soil via natural and (or) anthropogenic activities is another stress that crop plants regularly experience. The physical-geochemical properties of these heavy metals in soil and their subsequent uptake by plants have been found to adversely affect various physiological, morphological and biochemical processes of the plant, resulting in reduced crop productivity. Given the relevance of wheat as an important cereal across the globe, developing/screening wheat cultivars with low heavy metal accumulation promises reduced dietary exposure to heavy metals. To this effect, root transcriptomes of YM16, a low-Cadmium (Cd)-accumulating wheat genotype were analysed under cadmium-treated and untreated conditions [70]. Heavy metal contamination of the soil via natural and (or) anthropogenic activities is another stress that crop plants regularly experience. The physical-geochemical properties of these heavy metals in soil and their subsequent uptake by plants have been found to adversely affect various physiological, morphological and biochemical processes of the plant, resulting in reduced crop productivity. Given the relevance of wheat as an important cereal across the globe, developing/screening wheat cultivars with low heavy metal accumulation promises reduced dietary exposure to heavy metals. To this effect, root transcriptomes of YM16, a low-Cadmium (Cd)-accumulating wheat genotype were analysed under cadmium-treated and untreated conditions [134].
4. Metabolomic Approaches
Metabolomics is one of the most evolved and extensively explored omics technologies. It is an analytical profiling platform to measure and compare the metabolites present in biological samples at a given time. Metabolomics can be combined with high-throughput analytical chemistry and multivariate data analysis to unravel the molecular mechanisms at play in response to endogenous and exogenous environments of organisms. Its ability to efficiently complement other omics approaches makes it even more powerful for usage across all organisms [71]. In contrast to other omics approaches, metabolomics is efficient in tangibilizing the biochemical activities of apparent proteins and their subsequent metabolism into other proteins, making it rather easy to associate phenotypes [71]. Albeit not as extensively as transcriptomics, metabolomics has been employed in wheat to identify stress tolerance mechanisms and candidate genes by correlating the accumulation of metabolites in response to induced stresses [30][72][73]. An untargeted metabolomic analysis was employed to validate the use of wheat homeologous group 3 ditelosomic lines for identification and direct validation of genes regulating the accumulation of metabolites during the later stages of grain development [72]. Significant differences with respect to the accumulation of sugars were found between genetically modified wheat genotypes with their parental lines associated with the environment rather than the genotype [74]. Similar findings have also been reported in other investigations [73][75][76]. To assess the effects of nutrient deficiency on the metabolomic profile of wheat, a metabolomic approach in conjunction with transcriptomics was employed to assess the effects of nitrogen and sulphur deficiency on the remobilization of resources from a degrading canopy to developing grains in wheat [77]. The results obtained from both approaches corroborated, to suggest a suboptimal mobilization of N and (or) S supply to leaves but the supply to developing grains was always found to be optimum. A clever strategy employed by the plant was deciphered that utilizes the machinery in place for aminoacid biosynthesis to produce glutamine in developing grains. In the first seven days of seed development, a significant accumulation of glutamine was observed that was later converted to other amino acids and proteins over the subsequent 21 days of grain development [77]. The content of nitrogen and sulphur in the grains was found to increase at a steady rate of post-anthesis and nitrogen deficiency was found to adversely affect the accumulation of nitrogen and sulphur, suggesting that the availability of nitrogen at the vegetative stage determines the time and extent of the remobilization of resources [77]. Metabolomics is one of the most evolved and extensively explored omics technologies. It is an analytical profiling platform to measure and compare the metabolites present in biological samples at a given time. Metabolomics can be combined with high-throughput analytical chemistry and multivariate data analysis to unravel the molecular mechanisms at play in response to endogenous and exogenous environments of organisms. Its ability to efficiently complement other omics approaches makes it even more powerful for usage across all organisms [74]. In contrast to other omics approaches, metabolomics is efficient in tangibilizing the biochemical activities of apparent proteins and their subsequent metabolism into other proteins, making it rather easy to associate phenotypes [74]. Albeit not as extensively as transcriptomics, metabolomics has been employed in wheat to identify stress tolerance mechanisms and candidate genes by correlating the accumulation of metabolites in response to induced stresses [41,136,137]. An untargeted metabolomic analysis was employed to validate the use of wheat homeologous group 3 ditelosomic lines for identification and direct validation of genes regulating the accumulation of metabolites during the later stages of grain development [136]. Significant differences with respect to the accumulation of sugars were found between genetically modified wheat genotypes with their parental lines associated with the environment rather than the genotype [138]. Similar findings have also been reported in other investigations [137,139,140]. To assess the effects of nutrient deficiency on the metabolomic profile of wheat, a metabolomic approach in conjunction with transcriptomics was employed to assess the effects of nitrogen and sulphur deficiency on the remobilization of resources from a degrading canopy to developing grains in wheat [141]. The results obtained from both approaches corroborated, to suggest a suboptimal mobilization of N and (or) S supply to leaves but the supply to developing grains was always found to be optimum. A clever strategy employed by the plant was deciphered that utilizes the machinery in place for aminoacid biosynthesis to produce glutamine in developing grains. In the first seven days of seed development, a significant accumulation of glutamine was observed that was later converted to other amino acids and proteins over the subsequent 21 days of grain development [141]. The content of nitrogen and sulphur in the grains was found to increase at a steady rate of post-anthesis and nitrogen deficiency was found to adversely affect the accumulation of nitrogen and sulphur, suggesting that the availability of nitrogen at the vegetative stage determines the time and extent of the remobilization of resources [141].
An increased concentration of atmospheric CO2
has been reported to have an adverse effect on the metabolomic profile of the wheat plant. A change in 40 metabolites was found with an increase in atmospheric CO2 levels that was associated with the altered development of the plant [78]. This change was correlated with the decreased concentration of a few amino acids and derivatives that induce the raffinose synthesis pathway only during the vegetative phase but a reduced lysine turnover throughout the plant’s life cycle. Similarly, increased atmospheric CO levels that was associated with the altered development of the plant [143]. This change was correlated with the decreased concentration of a few amino acids and derivatives that induce the raffinose synthesis pathway only during the vegetative phase but a reduced lysine turnover throughout the plant’s life cycle. Similarly, increased atmospheric CO2 was found to be associated with a decrease in the accumulation of N-rich metabolites vis-a-vis a few organic acids and ribose-5-P [79]. was found to be associated with a decrease in the accumulation of N-rich metabolites vis-a-vis a few organic acids and ribose-5-P [144].
Furthermore, to assess the effect of Cd toxicity on wheat metabolome, the metabolome profile following Cd application was investigated from two hexaploid wheat genotypes, AK58, and ZM10, with a low and high Cd-accumulation in grains, respectively [80]. Compared to ZM10, AK58 was found to have a greater root antioxidant system and higher levels of Cd bound to root cell walls, owing to the increased accumulation of hemicellulose and pectin to aid Cd binding. To further the understanding of Cd toxicity, ameliorating effect of Boron (B) was analysed on wheat growth following exposure to Cd treatment [81]. It was found that plant growth under Cd stress was adversely affected and B application was not able to fully recover the plant [81]. However, following the B application, accumulated Cd and malondialdehyde levels in the shoot and root decreased significantly. In addition, B application led to a reduction in the activity of enzymes, such as SOD and peroxidase (POD), that were induced in response to Cd stress [81]. Furthermore, to assess the effect of Cd toxicity on wheat metabolome, the metabolome profile following Cd application was investigated from two hexaploid wheat genotypes, AK58, and ZM10, with a low and high Cd-accumulation in grains, respectively [148]. Compared to ZM10, AK58 was found to have a greater root antioxidant system and higher levels of Cd bound to root cell walls, owing to the increased accumulation of hemicellulose and pectin to aid Cd binding. To further the understanding of Cd toxicity, ameliorating effect of Boron (B) was analysed on wheat growth following exposure to Cd treatment [149]. It was found that plant growth under Cd stress was adversely affected and B application was not able to fully recover the plant [149]. However, following the B application, accumulated Cd and malondialdehyde levels in the shoot and root decreased significantly. In addition, B application led to a reduction in the activity of enzymes, such as SOD and peroxidase (POD), that were induced in response to Cd stress [149].
5. Proteomics Approaches
Proteins, along with their post-translational modifications, are crucial for plant stress responses. Proteomics studies, therefore, provide valuable information about the cellular pathways involved in stress adaptation and mitigation. Initially, the term ‘proteomics’ referred to the methods used to analyze numerous proteins at a time; however, the term has now expanded to include any approach that provides information on the abundance, properties, interactions, activities or structures of proteins in a sample [82]. The analysis of the protein profile of wheat plants in response to abiotic stresses, such as drought, salinity and heat, is well documented. Proteins, along with their post-translational modifications, are crucial for plant stress responses. Proteomics studies, therefore, provide valuable information about the cellular pathways involved in stress adaptation and mitigation. Initially, the term ‘proteomics’ referred to the methods used to analyze numerous proteins at a time; however, the term has now expanded to include any approach that provides information on the abundance, properties, interactions, activities or structures of proteins in a sample [150]. The analysis of the protein profile of wheat plants in response to abiotic stresses, such as drought, salinity and heat, is well documented.
Molecular mechanisms in response to drought were investigated in two wheat lines, Zhongmai 8601 and YW642, drought tolerance and Thinopyrum intermedium 7XL/7DS translocation line, respectively, under a drought stress environment [83]. Two-dimensional difference gel electrophoresis (2D-DIGE) was employed to explore the differential accumulation protein (DAP) after 20 days of post anthesis (DPA) and a total of 146 DAPs were identified [83]. Furthermore, MALDI-TOF/TOF-MS was employed to identify the 113 unique proteins connoted by these DAPs [83]. Of these, 48 unique proteins exhibited upregulation and were involved in plant stress response, protein metabolism and, energy metabolism. Most significantly, 14 DAP genes were reported to have high expression levels in the 7XL/7DS translocation line during grain development periods [83]. Of these, four genes were identified as responding to drought stress, of which two genes showed oxidoreductase and dehydrogenase activities. In addition, three genes with potential protein binding, catalytic and transmembrane transporter-type roles were identified under drought and heat stress [83]. Functional relevance of a plant growth-promoting rhizobacterium (PGPR) Enterobacter cloacae SBP-8 was assessed under excessive salinity (200 mM NaCl) stress by investigating proteome profiles in the bacterial-inoculated wheat plants with and without salt stress [84]. A total of 286 differentially expressed proteins (DEPs) were identified and the majority of them were linked to metabolic pathways, photosynthesis and stress mechanisms [84]. Furthermore, bacterial inoculation was found to upregulate the expression of the Hsp70, Hsp90 organizing protein and cold shock protein CS66 at 200 mM NaCl stress [84]. Molecular mechanisms in response to drought were investigated in two wheat lines, Zhongmai 8601 and YW642, drought tolerance and Thinopyrum intermedium 7XL/7DS translocation line, respectively, under a drought stress environment [158]. Two-dimensional difference gel electrophoresis (2D-DIGE) was employed to explore the differential accumulation protein (DAP) after 20 days of post anthesis (DPA) and a total of 146 DAPs were identified [158]. Furthermore, MALDI-TOF/TOF-MS was employed to identify the 113 unique proteins connoted by these DAPs [158]. Of these, 48 unique proteins exhibited upregulation and were involved in plant stress response, protein metabolism and, energy metabolism. Most significantly, 14 DAP genes were reported to have high expression levels in the 7XL/7DS translocation line during grain development periods [158]. Of these, four genes were identified as responding to drought stress, of which two genes showed oxidoreductase and dehydrogenase activities. In addition, three genes with potential protein binding, catalytic and transmembrane transporter-type roles were identified under drought and heat stress [158]. Functional relevance of a plant growth-promoting rhizobacterium (PGPR) Enterobacter cloacae SBP-8 was assessed under excessive salinity (200 mM NaCl) stress by investigating proteome profiles in the bacterial-inoculated wheat plants with and without salt stress [169]. A total of 286 differentially expressed proteins (DEPs) were identified and the majority of them were linked to metabolic pathways, photosynthesis and stress mechanisms [169]. Furthermore, bacterial inoculation was found to upregulate the expression of the Hsp70, Hsp90 organizing protein and cold shock protein CS66 at 200 mM NaCl stress [169].
To understand the effects of heat stress on wheat plants, proteome changes were analysed in the wheat kernel in the winter wheat cultivar Gaocheng 8901 under heat stress by iTRAQ (isobaric tags for relative and absolute quantitation) [85]. The iTRAQ analysis revealed quantitative information on 2493 proteins in the cultivar Gaocheng 8901 under heat stress, of which 116 were upregulated and 91 were downregulated [85]. A group of 78 DEPs were coupled to 83 KEGG signalling/metabolic pathways. Five DEPs, Elicitor responsive gene 3 (ERG3), brassinosteroid-insensitive 1 (BRI1), chaperone protein (CLPB1), histone cell cycle regulator (HIR1) and pre-mRNA processing factor (RSZ22), were involved in protein–protein interaction networks and were suggested to significantly impact the yield and quality of wheat grain under heat stress [85]. The relevance of stress-associated active proteins (SAAPs) involved in the process of the terminal heat tolerance of wheat has also been investigated [86]. The wheat To understand the effects of heat stress on wheat plants, proteome changes were analysed in the wheat kernel in the winter wheat cultivar Gaocheng 8901 under heat stress by iTRAQ (isobaric tags for relative and absolute quantitation) [161]. The iTRAQ analysis revealed quantitative information on 2493 proteins in the cultivar Gaocheng 8901 under heat stress, of which 116 were upregulated and 91 were downregulated [161]. A group of 78 DEPs were coupled to 83 KEGG signalling/metabolic pathways. Five DEPs, Elicitor responsive gene 3 (ERG3), brassinosteroid-insensitive 1 (BRI1), chaperone protein (CLPB1), histone cell cycle regulator (HIR1) and pre-mRNA processing factor (RSZ22), were involved in protein–protein interaction networks and were suggested to significantly impact the yield and quality of wheat grain under heat stress [161]. The relevance of stress-associated active proteins (SAAPs) involved in the process of the terminal heat tolerance of wheat has also been investigated [162]. The wheat cvs., HD2985 (heat-tolerant) and HD2329 (heat-sensitive) were studied under heat stress at 38 °C for 2 h to identify 4271 SAAPs by employing iTRAQ. Under heat stress, 2800 and 2225 differentially expressed SAAPs were upregulated and downregulated in the tolerant cultivar HD2985, while 800 and 3600 expressed SAAPs were upregulated and downregulated in the sensitive cultivar HD2329, respectively [86]. Using the gene ontology analysis, differentially expressed SAAPs were characterized into three majorly functional groups, namely, molecular functions (51%), biological processes (39%), and cellular components (10%) [86]. SAAPs have been classified to regulate in defence- and stress-related activities. However, expression of ribonuclease TUDOR-1, HSP90, HSP20, peroxidase and HSC70 has been found in the wheat , HD2985 (heat-tolerant) and HD2329 (heat-sensitive) were studied under heat stress at 38 °C for 2 h to identify 4271 SAAPs by employing iTRAQ. Under heat stress, 2800 and 2225 differentially expressed SAAPs were upregulated and downregulated in the tolerant cultivar HD2985, while 800 and 3600 expressed SAAPs were upregulated and downregulated in the sensitive cultivar HD2329, respectively [162]. Using the gene ontology analysis, differentially expressed SAAPs were characterized into three majorly functional groups, namely, molecular functions (51%), biological processes (39%), and cellular components (10%) [162]. SAAPs have been classified to regulate in defence- and stress-related activities. However, expression of ribonuclease TUDOR-1, HSP90, HSP20, peroxidase and HSC70 has been found in the wheat cv
. HD2985 (heat-tolerant), and downregulation was observed in the wheat cv. HD2329 (heat-susceptible) under heat stress [86].
. HD2329 (heat-susceptible) under heat stress [162].
6. Multiomics Approaches
Since the advancements in omics technologies and computational tools, the use of a multiomics approach has become a major area of thrust to answer burning questions in the stress biology of a crop and to reduce the number of false positives arising from the use of a single data
[87][88][172,173]. It has been proposed that, by inspecting the change in correlation in the transcript–protein–metabolite between the control and stress conditions, biological processes strongly regulated by the plants can be recognised.
Various web-based tools and visualization portals are available to analyze the multi-omics datasets, such as PAINTOMICS, KaPPA-view, COVAIN, and O-miner
[89][90][91][92][93][174,175,176,177,178]. In PAINTOMICS, the integrated visualization of transcriptomics and metabolomics datasets is possible and displays the data on KEGG pathway maps. The KaPPA-view tool allows the integration of transcript and metabolite data on plant metabolic pathway maps. The COVAIN tool offers statistical analysis of the integrated omics dataset through the KEGG pathway and gene ontology analysis
[92][177].
Both proteomics and metabolomics approaches were employed for two spring-wheat cultivars, Bahar and Kavir, drought-tolerant and drought-susceptible, respectively, to understand the underlying biochemical networks at play in wheat leaves under drought stress
[94][154]. Metabolomic analysis revealed that the levels of primary metabolites, such as amino acids, sugars and organic acids, were found to change in response to water deficiency
[94][154]. In the Bahar cv, the accumulation of branched-chain amino acids, lysine, proline, aromatic, arginine and methionine was found in response to drought stress, in addition to the activation of shikimate pathway-mediated tryptophan accumulation aiding auxin production
[94][154]. In the Kavir metabolome, only two pathways were found to be significantly affected in response to stress, one being of purine metabolism
[94][154]. Only a few alterations in the metabolomic profile of Kavir were potent enough to induce susceptibility to drought stress, suggesting that unravelling the subtle changes in complex pathways leads to profound phenotypical changes
[94][154].
Combined metabolomic and proteomic approaches was employed to dissect the resistance mechanism conferred by Fhb1 QTL in the near-isogenic lines (NILs) derived from the wheat genotype Nyubai
[95][142]. The comparison of the metabolomic and proteomic profiles in the NILs revealed that the shunt phenylpropanoid pathway-producing metabolites, such as hydroxycinnamic acid amides, phenolic glucosides and flavonoids, played an important role
[95][142]. Using confocal microscopy, it was confirmed that cell wall thickening, due to the deposition of hydroxycinnamic acid amides, phenolic glucosides and flavonoids, was responsible for imparting resistance rather than the conversion of DON to less toxic deoxynivalenol 3-O-glucoside, demonstrating alternate novel pathways that could play a pivotal role
[95][142].
Large-scale multi-omics analysis was used to dissect the wheat stem solidness and resistance to wheat stem sawfly (WSS)
[96][179]. A combined transcriptomic, metabolomic and proteomic approach was deployed on two wheat cultivars, the solid-stemmed Choteau and semi-solid-stemmed Scholar, differentiating for a QTL identified previously on chromosome 3B for solid stem
[96][179]. The semi-solid-stemmed cultivar showed a differential regulation of 15 transcripts on WSS infection, of which 5 were upregulated and coded for an auxin efflux carrier component, pathogenesis-related (PR) genes 5 (CPR-5) protein, NADH dehydrogenase (NDH-A) and a magnesium transporter
[96][179]. The solid-stemmed Choteau variety, on the other hand, showed nine DEGs, of which only one was found to be upregulated, while the remaining transcripts were downregulated
[96][179].
Molecular mechanisms underlying the adaptation of wheat were determined in response to potassium (K) deficiency by investigating the transcriptomes and metabolomes of a panel of wheat accessions differing in K-deficiency tolerance
[97][181]. The panel was subjected to a low-K treatment under hydroponic culture conditions for 14 days and root samples were collected for transcriptomic and metabolomic analyses
[97][181]. It was found that the three CIPK (serine/threonine protein kinases)-encoding DEGs, i.e., CIPK14, CIPK9 and CIPK27, were upregulated in KN9204 and unchanged in BN207, while the expression of the three other DEGs (CIPK19, CIPK15 and CIPK29) was downregulated in BN207. These CIPKs were suggested to be underlying candidates for low-K tolerance. Recently, a multiomics approach was employed to identify the genes conferring a dense spike in a wheat-dense spike mutant (wds) obtained from a landrace Huangfangzhu
[98][128]. Two large deletion segments on chromosome 6B, at 334.8–424.3 and 579.4–717.8 Mb, in the wds mutant and 499 genes were identified within the deleted regions
[98][128].
In most of the GWAS studies, the biological mechanisms underlying the associations are unknown. To deal with this, GWAS has been integrated with metabolite profiling and/or gene regulatory network or pathway analysis and this approach has proven to be very effective in increasing the utility of information gained from GWAS
[99][100][101][183,184,185]. A combination of GWAS and SNP-phenotype network approaches was employed in a core collection of wheat to identify the genetic basis of grain yield and spikelet-fertility-related traits
[101][185]. The 118 SNPs that were found to be significant in their study at a false discovery rate (FDR) = 0.01 were used in the genotype–phenotype network analysis, of which 14 SNPs directly interacted with many of the agronomic traits. In silico analysis of these 14 SNPs revealed the strong involvement of metal ion transport and Gibberellin 2-oxidases (GA2oxs) genes in controlling the spikelet sterility
[101][185].