Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ekaterina Belova and Version 2 by Camila Xu.
Microsatellite instability (MSI) occurs in a wide variety of tumor types and is one of the most important predictive biomarkers for immune checkpoint inhibitor therapy.
- microsatellite instability
- mismatch repair
- biomarker
1. Introduction
Microsatellite instability (MSI) results from impaired DNA mismatch repair (MMR) and causes an accumulation of mutations in microsatellites (MS), also called short tandem repeats (STRs). STRs consist of repeated sequences of 1–6 nucleotides and account for 3% of the genome, both coding and noncoding regions [1][2][3][1,2,3]. The nature of MS determines its outstanding tendency to accumulate errors. It happens due to DNA slippage in the process of DNA replication, which usually leads to a change in MS length [4][5][4,5].
The MMR system is highly conserved across species. MMR is responsible for the recognition and correction of mismatched nucleotides and plays a key role in maintaining genomic stability [6][7][8][6,7,8]. Four major proteins encoded by the MLH1 (mutL homologue 1) [9][10][9,10], MSH2 (mutS homologue 2) [11], MSH6 (mutS homologue 6), and PMS2 (postmeiotic segregation increased 2) [12] genes play a central role in this process [13]. The MMR system functions through the formation of heterodimers (hMutS and hMutL). hMutS consists of MSH2 and one of the secondary proteins, either MSH6 or MSH3, and recognizes mismatched nucleotides and small indels [7][8][7,8]. The hMutL heterodimers, consisting of MLH1 and one of the secondary proteins, PMS2, PMS1, or MLH3, participate in MMR reactions [14][15][16][14,15,16] via its endonuclease activity [17], 5′ nicking [18], modulation, and termination of exonuclease 1′s (Exo1) activity [18][19][20][18,19,20]. Thus, hMutL deficiency leads to Exo1 hyperactivity and increased DNA excision [19][20][19,20]. The inactivation of at least one of the following genes: MLH1, MSH2, MSH6, or PMS2, due to germline and/or somatic mutations or epigenetic silencing, results in the MMR system deficiency (dMMR) [21][22][21,22].
MSI occurs among various tumor types, and is most common in colorectal, small bowel, endometrial, and gastric cancers [3][23][24][25][26][27][28][29][3,23,24,25,26,27,28,29]. Most cases of MSI are sporadic, arising from epigenetic inactivation of MLH1 gene expression. However, the MSI can also be caused by Lynch syndrome, a hereditary condition resulting from germline pathogenic mutations in the MMR genes, coupled with inactivation of the second allele [30][31][32][30,31,32].
MSI is the one of the major biomarkers predictive of the immune checkpoint inhibitor (ICI) benefit across cancer types, both in Lynch syndrome-related and sporadic tumors. ICI therapy aims to overcome tumor immune escape through targeting immune inhibitory molecules (e.g., PD-1, PD-L1, LAG3, and CTLA4) expressed on the surfaces of tumor and immune cells [33][34][35][33,34,35]. Correct assessment of MSI is critical for adequate therapeutic decisions. According to ESMO recommendations [30], several methods are used in clinical practice to assess MSI status. The IHC method indirectly assesses MSI by detecting loss of staining for MLH1, MSH2, PMS2, and MSH6 proteins. An advantage of using IHC to detect MMR proteins is its convenience and ability to identify the target gene for future mutational confirmation [24]. PCR-based approaches directly determine MSI status via amplification of specific microsatellite repeats. ESMO suggests PCR in case of indeterminate IHC results, including disagreement or difficulties in interpreting IHC. The five poly-A mononucleotide repeats panel (BAT-25, BAT-26, NR-21, NR-24, NR-27) is considered the current standard [30], though the Bethesda panel, comprising of two mononucleotide (BAT-25 and BAT-26) and three dinucleotide (D5S346, D2S123, and D17S250) repeats, is also widely utilized in clinical practice [36][37][36,37]. In diagnostics, MSI (previously known as MSI-H) denotes alterations in the lengths of several MS (e.g., 2 of 5 loci in a standard PCR test with five poly-A mononucleotide repeats). In contrast, if the number of unstable loci does not exceed one, it is termed as MSS (microsatellite stable). NGS-based MSI detection is considered as one of the most promising, since its advantages include higher accuracy and an expanded spectrum of microsatellites analyzed, which is relevant for non-CRC tumors that can harbor a non-standard set of unstable microsatellites [38]. NGS also allows simultaneous analysis of a comprehensive spectrum of clinically significant biomarkers [14][30][14,30].
The first FDA-approved ICI drug was ipilimumab, which was initially approved in 2011 for the treatment of melanoma and is now used in a limited range of cancers. Pembrolizumab is a second FDA-approved ICI, which has the widest range of indications, including tissue-agnostic indications [39]. To date, more than 6 ICI have been approved for site-specific or tumor-agnostic indications. Overall, the objective response rate (ORR) for tumors with MSI varies between 34% and 69% depending on the line of therapy, while the rate of pathologic complete response (pCR) may reach 100% in neoadjuvant settings, which indicates a high efficacy of checkpoint inhibitors in MSI tumors [40][41][42][40,41,42]. At the same time, about a quarter of metastatic colorectal cancers and up to half of some other types of cancer are intrinsically resistant to ICI [43][44][43,44]. Several mechanisms of such an intrinsic resistance have been proposed, including the activating mutations in the RAS/RAF signaling pathway [45], mutations in the antigen presentation machinery and interferon pathway genes [46][47][46,47], establishment of an immunosuppressive microenvironment [48], as well as the influence of the gut microbiome [49] and immunoediting theory [50].
2. MSI: Molecular Epidemiology across Cancer Types
The MSI phenotype can be found in many cancer types. In a study analyzing more than 11,000 tissue samples from patients with 39 cancer types, MSI was found in 27 tumor types (overall in 3.8% of all samples) [51][83]. The tumor types where the MSI phenotype is observed include colon, gastric, endometrial, ovarian, hepatobiliary tract, urinary tract, brain, and skin cancers (Figure 12A). Of those, the highest prevalence of MSI is in colorectal cancer (10.2%, range 6.6–14.5%) [23][24][30][52][53][54][55][56][57][58][59][23,24,30,84,85,86,87,88,89,90,91], endometrial cancer (especially endometrioid histotype) (21.9%, range 15.1–29.6%) [25][26][27][52][60][61][62][25,26,27,84,92,93,94], gastric cancer (8.5%, range 6.4–10.9%) [3][27][28][30][52][63][3,27,28,30,84,95], and small bowel cancer (14.3%, range 5.4–26.3%) [52][84]. In gastric cancer, the frequency of MSI/dMMR varies significantly within histological subtypes: from 0.9% in the mixed-type and 2.9% in the diffuse-type, to 10.7% of the intestinal-type [28][64][28,96]. In other types of cancer, the MSI rate is relatively low [52][84], specifically 2–10% in ovarian [30][65][66][67][30,60,97,98] and only 1–2% in pancreatic cancer [30][68][69][70][30,99,100,101]. The assessments of the MSI rate in urothelial carcinoma are highly contradictory, with the reported values ranging from 1% to as high as 46% [71][72][73][102,103,104]. The MSI phenotype is also reported in Lynch syndrome-unrelated cancer types—in glioblastoma, cervical cancer, small intestine, melanoma, sarcoma, and others, but in these cancer types, MSI is much rarer [30][74][75][30,105,106].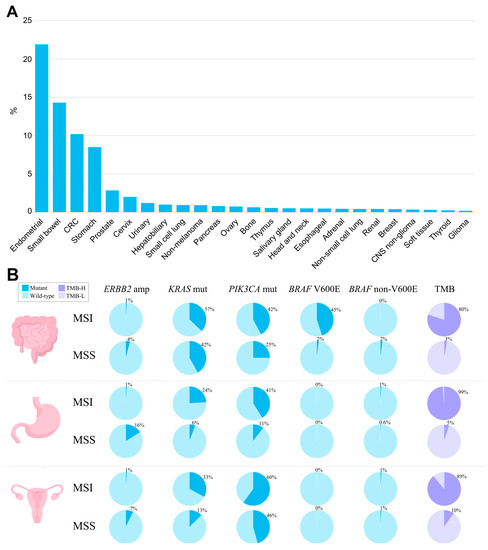
Figure 12. The landscape of MSI and genomic characteristics of MSI tumors across cancer types, according to TCGA. (A) Prevalence of MSI (%) across tumor types. (B) Prevalence of genomic alterations typically found in MSI and MSS tumors in colorectal adenocarcinoma, stomach adenocarcinoma, and uterine corpus endometrial carcinoma.