Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Adriano Duatti.
The β− emitter, rhenium-188 (188Re), has long been recognized as an attractive candidate for targeted cancer radionuclide therapy (TRNT). This transition metal shares chemical similarities with its congener element technetium, whose nuclear isomer technetium-99m (99mTc) is the current workhorse of diagnostic nuclear medicine. This pair of elements forms a characteristic matched pair for chemical and medical research and applications.
- rhenium
- transition metal chemistry
- ligand field theory
- frontier orbitals
- nuclear medicine
- <
- math xmlns="http://www.w3.org/1998/Math/MathML" display="block">
- <
- semantics>
- <
- mrow>
- <
- mo>
- &
- /mo>
- mi>
- G<
- /mi>
- =&l
1. Introduction
At the end of the last century, the radioisotope rhenium-188 (188Re) was among the first radionuclides to be proposed for the development of therapeutic radiopharmaceuticals [1]. There were several reasons to believe that 188Re was an appropriate choice for medical applications. The nuclear properties of 188Re are extremely interesting. It decays through the emission of β− particles with mean energy Emean = 763 keV and maximum energy Emax = 2.12 MeV which is advantageous for the delivery of cytotoxic radiation to the tumour. The emission of a γ photon, with energy Eγ = 155.04 keV (15.05%), is also associated with the β− decay. This isomeric transition is almost ideal for applying single photon emission tomography (SPECT) and outperforms emissions from other β− emitting radionuclides in terms of resolution for dosimetry purposes [2]. The relatively short half-life of 16.98 h when combined with the very high specific activity of this radionuclide can become an additional advantage for some specific clinical applications [3,4][3][4].
An attractive feature that initially contributed to stimulating the interest in 188Re was the similarity between the chemical properties of the two congener elements rhenium and technetium [5,6,7][5][6][7]. The reasoning was that 188Re radiopharmaceuticals could be produced simply by replacing the radionuclide 99mTc with the radionuclide 188Re in the well-established reaction scheme used for the preparation of 99mTc radiopharmaceuticals. In the periodic table, technetium and rhenium belong to Group 7, and are categorized in the second and third row of the transition metal series, respectively. A variety of radiopharmaceuticals labelled with the metastable γ emitting nuclear isomer 99mTc for the diagnosis of many pathological conditions has been investigated [1,5][1][5]. This was accomplished by the development of efficient chemical methods for the synthesis of 99mTc complexes in physiological solutions and in sub-millimolar concentrations (also called ‘no-carrier-added level’). A prerequisite for clinical translation was safety and stability during intravenous administration in vivo. The development of highly efficient labelling methods stemmed from the richness of the chemical properties of the element technetium. This unique characteristic of Group 7 transition metals allowed the production of a multitude of technetium complexes characterized by a wide variety of molecular structures.
Similarly, it was argued that the same structural richness could be observed for 188Re radiopharmaceuticals which ultimately could be used as therapeutic agents with almost no need for optimization of labelling methods. This chemical parallelism between the two radioisotopes was interpreted as one of the earliest examples of ‘theranostic pair’ (although the term ‘matched pair’ was originally used), a ubiquitous concept in current radiopharmaceutical research [9][8]. Nature is not always respectful of the elegant symmetries described in scientists’ theoretical models and usually deviates from the expected results. In the example of the congener elements rhenium and technetium, the chemical similarities between the two elements are only superficial, and this diversity becomes more pronounced when studying their chemical reactivity at the no-carrier-added level as implemented in radiopharmaceutical production. This forced the researchers to abandon the hope that 188Re radiopharmaceuticals could be produced simply by replacing the nuclide 99mTc with the nuclide 188Re in the well-established reactions utilized for the preparation of 99mTc radiopharmaceuticals. Commonly, when this approach was naively attempted, the production of the corresponding 188Re radiopharmaceuticals failed dramatically. Therefore, it became clear that it was necessary to develop specific labelling methods for preparing 188Re radiopharmaceuticals and that the methods used for the preparation of 99mTc radiopharmaceuticals were simply not superimposable [9,10][8][9].
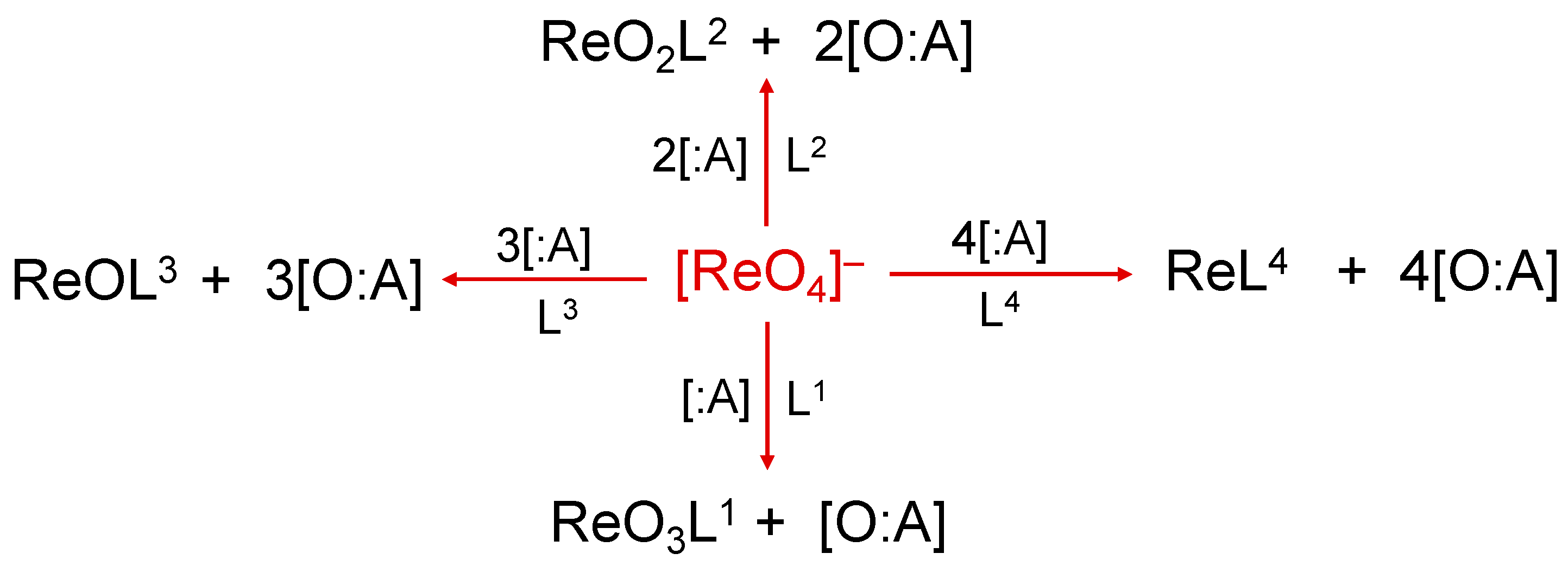
2. The Reduction of the Perrhenate Anion [188Re][ReO4]−
The radioisotope 188Re is produced from the weak β− decay of the parent nuclide tungsten-188 (188W). The parent nuclide is in turn obtained by double neutron capture of a 186W target after neutron irradiation in a high-flux nuclear reactor according to the reaction pathway 186W + 2n → 188W → 188Re + β− + ν¯ (where n = neutron and ν¯ = electron antineutrino). The produced 188W can then be loaded onto a generator system to afford a long-term source of no-carrier added 188Re. This makes the use of 188Re in therapeutic applications even more fortuitous [12,13,14][10][11][12]. The analogous 188W/188Re generator follows the same design strategy utilized for the development of the 99Mo/99mTc generator where the long-lived 188W parent nuclide (69.4 days) is absorbed on an alumina column. This system can be periodically eluted with a sterile saline solution (0.9% NaCl) that allows the selective separation of 188Re, in the chemical form of sodium perrhenate ([188Re]ReO4Na), in very high specific activity [12,13,14][10][11][12]. Similarly to the pertechnetate anion, [99mTc][TcO4]−, the perrhenate anion, [188Re][ReO4]−, is the universal starting material for the preparation of 188Re radiopharmaceuticals. The two tetraoxo anions, [MO4]− (M = Re, Tc), share the same molecular structure where the metal is bound to four oxygen atoms in a tetrahedral arrangement. Consequently, the preparation of new 188Re compounds necessarily requires the removal of at least one, or ultimately, all four of the oxygen atoms from the perrhenate anion. This process can be accomplished through the reaction of [188Re][ReO4]− with an appropriate chemical species (:A) able to accept oxygen atoms. A schematic, step-by-step representation of this reaction is reported in Figure 1.Figure 1. Oxygen atom transfer reactions using [188Re]perrhenate as starting substrate. The symbol ‘:A’ represents the oxygen-atom acceptor and Ln (n = 1–4) the ligand (or multiple ligands) suitable to form a stable complex with one of the various intermediate Re-oxo forms. Examples of :A are SnCl2 or tertiary phosphines.
Traditionally, the overall reaction scheme depicted in Figure 1 is called the reduction of the [188Re][ReO4]− anion and this definition originates from a different description of the oxo-transfer reaction based on the concept of ‘oxidation state’. The attribution of a positive or negative number (usually an integer number) to each atom of a particular element when bound to other atoms inside a molecule is a very old procedure which, nonetheless, still provides a very convenient method to classify the various chemical properties of a single element. The oxidation states of an element are purely formal numbers as they cannot be derived from a more fundamental theory. On the contrary, they are calculated following an abstract set of rules (that can be conveniently modified or extended when necessary). To simplify here, considering that oxygen and hydrogen play a dominant role on a planet covered by water, the oxidation states −2 and +1 are arbitrary attributed to these two elements, respectively, when they form a molecule. The other key assumption necessary to calculate the oxidation states is that the sum of all oxidation states of all elements in a molecule should be equal to the overall net charge of that molecule. Based on these simple rules, it is straightforward to calculate the oxidation state of the rhenium atom in the [188Re][ReO4]− anion. Since this molecular anion carries an overall negative charge of –1, and the rhenium metal is bound to four oxygen atoms, the metallic oxidation state, x, should satisfy the following equation: x + [4 × (−2)] = –1 resulting in x = +7. In the reaction diagram of Figure 1, assuming that the contribution to net charge of the molecule by the ligands Ln (n = 1–4) is zero, it follows that the removal of each oxygen atom by the acceptor :A necessarily decreases the oxidation state of rhenium by a factor 2 as, in the final sum of the overall oxidation states, one oxygen atom is missed. This means that for each oxygen-atom transfer, the oxidation state of rhenium is progressively reduced from +7 to a lower oxidation state (a process called ‘reduction’). In conclusion, the oxygen-atom transfer reaction starting from [188Re][ReO4]− can also be also viewed as a ‘reduction of the perrhenate anion’. Obviously, when there exists a non-zero contribution by the ligand Ln to the overall sum of the oxidation states, the final oxidation state of rhenium should be calculated accordingly.
3. The Expansion of the Coordination Sphere
The electrochemical potential for a redox reaction (ΔE) is regulated by the Nernst equation, which in turn depends on the difference between the standard reduction potential of the reductant and that of the oxidant (∆E°), the absolute temperature (T), and the logarithm of the concentration ratio (Q) between products and reactants:
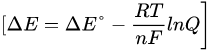
where n is the number of moles of electrons exchanged in the reaction and F is the Faraday constant). Since the impact of the logarithmic term on the value of ΔE is less than 10%, it is arduous to increase the yield of a redox process simply acting on the macroscopic variables such as temperature and concentration. Therefore, other chemical approaches should be employed to raise the radiochemical yield for the reduction of [188Re][ReO4]−. Some insight on a possible strategy can be obtained by considering the thermodynamic expression of the electrochemical potential:
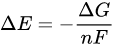
where ΔG is the variation of the free energy of the reaction and n is still the number of moles of electrons exchanged in the redox process and F the Faraday’s constant. In turn,
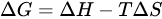
where ΔH is the variation of enthalpy, or of the heat of reaction at constant pressure, and ΔS is the change in entropy. Obviously, for a given reaction, the formation of products is favoured when the value of ΔG is negative. A negative value of ΔH can be obtained only through the formation of more stable chemical bonds in product molecules, or by acting on the temperature. However, under normal conditions, these factors alone cannot bring about a significant change of the free energy, and consequently, of the electrochemical potential ΔE, necessary to increase the yield for the reduction of [188Re][ReO4]−.
The remaining factor left for modification in the expression of ΔG is the variation of entropy ΔS. The change in entropy should be largely positive to ensure the negativity of ΔG. In the language of statistical thermodynamics, an increase in entropy entails a concomitant increase of the degrees of freedom in going from reactants to products. Here, the initial system is [188Re][ReO4]−, which is a molecular ion having a tetrahedral geometry. An increase in entropy occurs only if the tetrahedral, four-coordination arrangement around the central rhenium atom can be expanded to achieve a higher coordination number, namely, five-, six, or higher. This expansion is necessarily linked to a corresponding change of the molecular geometry from tetrahedral to higher geometries (for example, square-pyramidal, octahedral, and dodecahedral).
4. Basic Rhenium Coordination Chemistry
In organic chemistry, the notion of ‘functional group’ is a very useful approach for describing the chemical properties of an organic molecule. Using this approach, the structure of an organic molecule is viewed as made up of different molecular pieces each corresponding to a particular grouping of atoms. For example, the group of atoms composed by a nitrogen atom and two hydrogen atoms (NH2) constitutes the amino functional group and its presence imparts characteristic structural properties and reactivity to the hosting molecule. A similar approach can be also applied to rhenium chemistry and, consequently, several ‘inorganic functional groups’ (also called ‘cores’ or ‘metallic fragments’) have been identified. Likewise with the organic counterparts, an inorganic functional group governs the chemical behaviour of a specific class of metallic complexes and determine their structural properties. Some of the most important rhenium functional groups are the oxygenated cores Re(VII) trioxo {[Re(O)3]+}, Re(V) dioxo {[Re(O)2]+}, and Re(V) oxo {[Re(O)]3+}, the nitrido core {[Re(V)N]2+}, the diazenido core {[Re(V)(NNR)]2+} (where R is a terminal substituent), and the tris-carbonyl core {[Re(CO)3]+} [6,8,25,26][6][13][14][15]. A reproducible procedure for the high-yield preparation of 188Re radiopharmaceuticals from generator-produced [188Re][ReO4]− has been successfully achieved only with a few cores. Specifically, 188Re-radiopharmaceuticals bearing the Re(V) oxo and Re(V) nitrido cores have been produced almost exclusively at tracer level and evaluated in human studies [4,27][4][16]. Exceptions are the 188Re radiopharmaceuticals [188Re][Re(PhCS3)2(PhCS2)] (Ph = phenyl group), which has been introduced for the treatment of liver cancer [3,28][3][17] and incorporates the [ReIII(S2)(S3)2] functional moiety, and the polymeric 188Re bisphosphonate agents of unknown structure used for bone-pain palliation [29,30][18][19]. Attempts to produce 188Re radiopharmaceuticals using other cores have been always plagued by poor radiochemical yields and low stability under physiological conditions.5. Chemical Differences between the Re(N) and Re(O) Cores
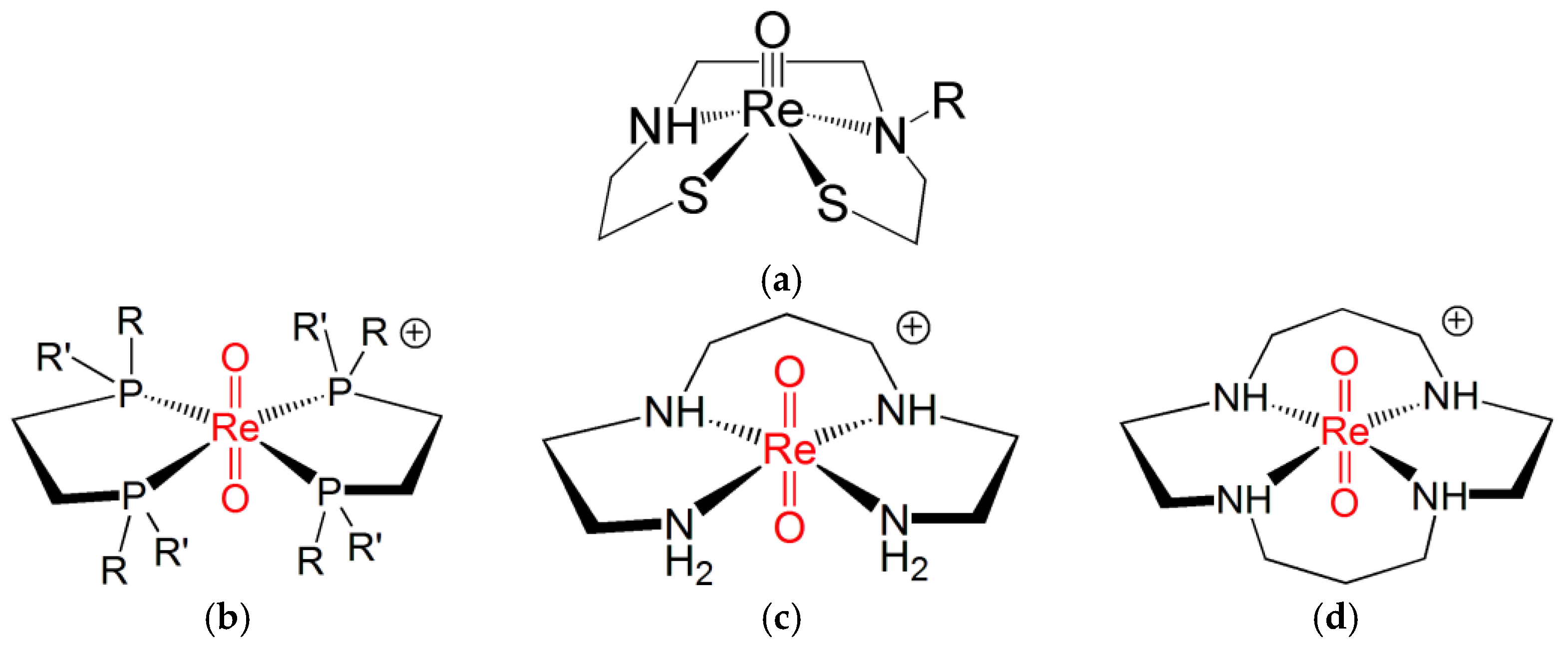
Despite the similarity between the FOs diagrams of Re(N) and Re(O) functional groups, there exist marked differences in their chemical behaviour. For example, the remarkable property of complexes containing the Re(V) nitrido group to switch between sp and tbp geometries upon coordination by π-acceptor ligands is not displayed by Re(V) oxo complexes. Conversely, the binding of π-acceptor ligands to the [Re≡O]3+ moiety invariably forces the coordination arrangement to turn into octahedral geometry [45][20]. This geometrical change from square-planar to octahedral coordination may trigger the formation of the Re(V) dioxo group, trans-[O = Re = O]+, where a second oxygen atom binds the [Re≡O]3+ group in trans position to the oxo atom. Usually, the second oxygen atom originates from a doubly deprotonated water molecule after its nucleophilic attack to the Re(V) centre. If the trans-[O = Re = O]+ group is not sufficiently stabilized by some appropriate ligand, this event may initiate the subsequent chain of hydrolytic attacks to the Re(V) ion, which finally leads to the formation of the perrhenate anion [ReO4]−. Ligands capable of stabilizing the trans-[O = Re = O]+ core are bidentate diphosphines [R(R′)–P–P–(R′)R where R, R′ = organic substituents], ethylenediamine, and acyclic and cyclic tetramines. Figure 72 illustrates a few examples of octahedral complexes comprising the trans-[O=Re=O]+ moiety [6,7,8,22,25,46][6][7][13][14][21][22].Figure 72. The molecular structures of (a) an example of sp Re(V) oxo complex and of octahedral trans)-dioxo Re(V) complexes with (b) diphosphines and (c) acyclic and (d) cyclic tetramines ligands (R, R′ = any pendant organic substituents) [6,7,8,25,46,47][6][7][13][14][22][23].
The chemical factors that are usually invoked to explain the chemical differences between the [Re≡N]2+ and [Re≡O]3+ functional groups are the lower electronegativity and smaller atomic radius of the nitrogen atom with respect to the oxygen atom. This allows a more extensive overlap of the valence AOs of Re and N, and the formation of a strong Re≡N multiple bond [48][24]. Notably, in many Re(V) oxo octahedral complexes, the Re(V) oxo bond distance is closer to that of a Re=O double bond than to that of a Re≡O triple bond predicted by the FOs diagram reported in Figure 3.
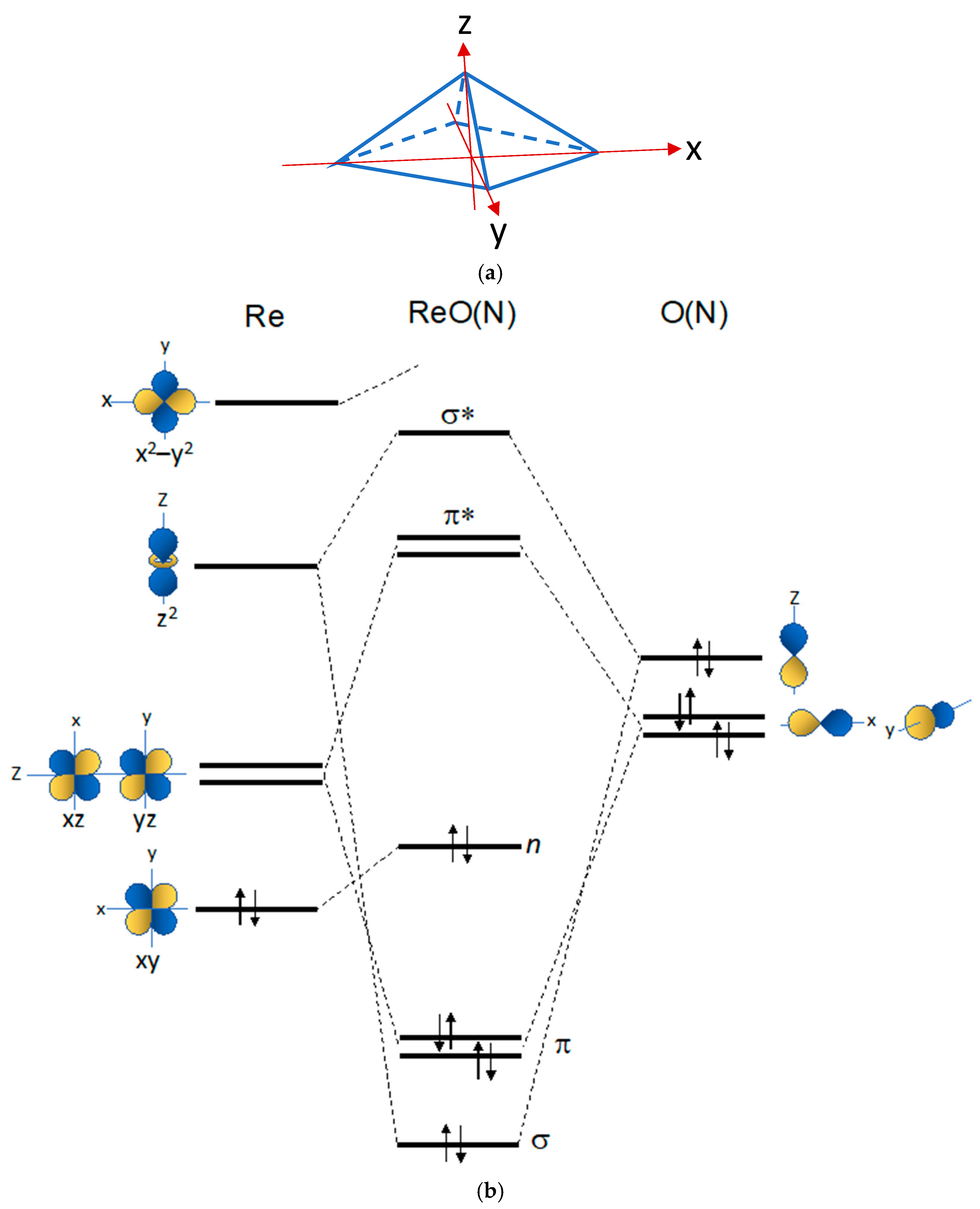
Notably, in many Re(V) oxo octahedral complexes, the Re(V) oxo bond distance is closer to that of a Re=O double bond than to that of a Re≡O triple bond predicted by the FOs diagram reported in Figure 3.
In sp Re(V)-nitrido complexes, the shorter Re≡N bond length determines a strong distortion of their square pyramidal geometry that generate their characteristic umbrella-shaped structure. This geometrical distortion causes the metal atom to move up from the square plane towards the nitride nitrogen atom with the concomitant bending of the four coordination bonds. This effect can efficiently shield the Re(V) atom from the nucleophilic attack of an incoming water molecule in the trans position of the nitrido nitrogen atom. Therefore, this geometrical distortion can reasonably explain the higher inertness of Re(V) nitrido sp complexes against hydrolysis by water molecules [18,33,48,49,50,51][24][25][26][27][28][29]. One example of Re(V) nitrido complexes with a pseudo tbp geometry is illustrated in Figure 84a, while Figure 84b shows the structure of a bis-substituted dithiocarbamate complex with a distorted sp geometry. It is worth noting here that, for the sake of simplicity, in Figure 84a, D3h symmetry has been ideally (and arbitrarily) attributed to the complex [Re(N)Cl2(PPh3)2] whereas its real geometry is distorted by the sterically encumbering PPh3 substituents and better described by a C2v symmetry.
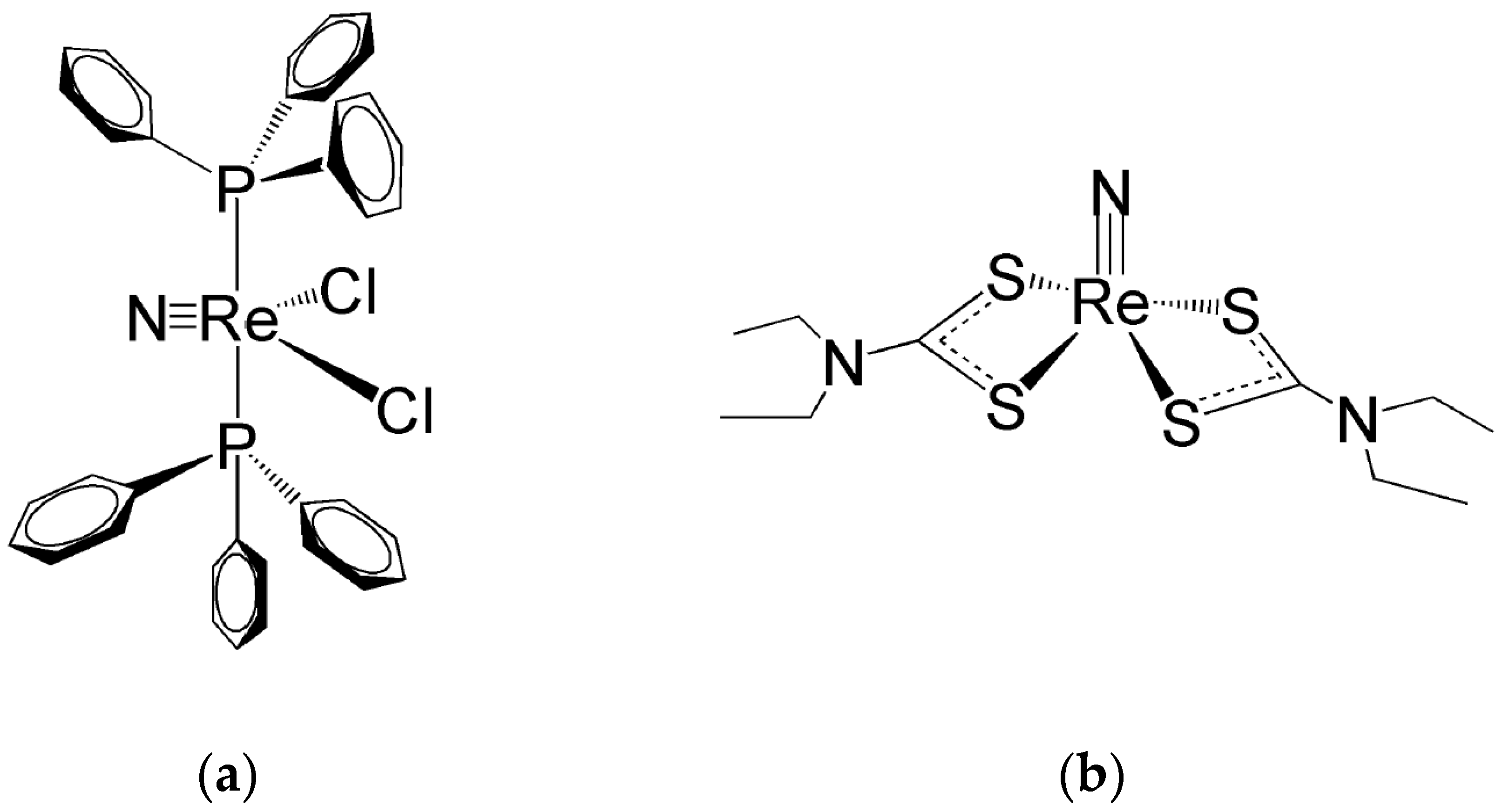
Figure 8. (a) The mixed halogeno-phosphino Re(V) complex (Re(N)Cl2(PPh3)2) [33][26] and (b) the bis-substituted complex (Re(N)(CS2NEt2)2) [51][29].
6. Matching the Nuclear and Biological Properties of 188Re Radiopharmaceuticals
To translate 188Re as a therapeutic radionuclide to the clinic, a robust production method needs to be in place. Inspired by the chemical similarities between rhenium and technetium, many research groups attempted to use the kit formulations developed for the preparation of 99mTc radiopharmaceuticals to produce the corresponding 188Re radiopharmaceuticals. However, when studied at the tracer level, the predicted chemical similarities were found to be more elusive than expected. As mentioned previously, only a small number of 188Re radiopharmaceuticals have been successfully introduced into the clinical use using freeze-dried kit formulations. The design of these radiopharmaceuticals was almost invariably based on the chemistry of Re(V) oxo and Re(V) nitrido cores and of 188Re complexes containing the sulphur rich ReIII(S3)2(S2) metallic group [3,4,18,25,27,28][3][4][14][16][17][25].
Indeed, it is becoming increasingly important to obtain an adequate match between the nuclear characteristics of the radionuclide, the pharmaceutical carrier, and the proposed biological target to elicit the most powerful therapeutic effect on the disease profile. This strategy can also favour a higher throughput from development to clinical application. The high-energy and relatively short-lived β− emission of 188Re suggests that high-capacity target sites could be more suitable for therapeutic applications based on this radionuclide. Current research on targeted radionuclide therapy is mostly focused on selecting interactions of the therapeutic agent with biological substrates expressing low-capacity receptors. These sites should be more appropriate for radionuclides emitting particles with high linear energy transfer (LET). This category includes α (e.g., terbium-149, astatine-211) and Auger emitters (e.g., iodine-125, copper-64). It is worth noting that a critical issue related to these receptor-specific therapeutic radiopharmaceuticals is that their uptake should always occur in proximity to the cell nucleus to ignite the disruption of the cancerous cell. For this approach, therefore, a strong binding to the receptor followed by an efficient internalization of the radiolabelled vector are necessary prerequisites. Conversely, rather than focusing on receptor interactions and internalization, 188Re-based radiopharmaceuticals should be designed to target high-capacity sites that commonly allow a rapid and extensive accumulation of the cytotoxic payload at the tumour site associated with a fast clearance from background tissues.
References
- Dilworth, J.R.; Parrott, S.J. The biomedical chemistry of technetium and rhenium. Chem. Soc. Rev. 1998, 27, 43–55.
- Esquinas, P.L.; Rodríguez-Rodríguez, C.; Carlos De La Vega, J.; Bokharaei, M.; Saatchi, K.; Shirmohammad, M.; Häfeli, U.O.; Sossi, V.; Celler, A. 188Re image performance assessment using small animal multi-pinhole SPECT/PET/CT system. Phys. Med. 2017, 33, 26–37.
- Lepareur, N.; Lacœuille, F.; Bouvry, C.; Hindré, F.; Garcion, E.; Chérel, M.; Noiret, N.; Etienne Garin, E.; Knapp, F.F.R., Jr. Rhenium-188 labeled radiopharmaceuticals: Current clinical applications in oncology and promising perspectives. Front. Med. 2019, 6, 132.
- Uccelli, L.; Martini, P.; Urso, L.; Ghirardi, T.; Marvelli, L.; Cittanti, C.; Carnevale, A.; Giganti, M.; Bartolomei, M.; Boschi, A. Rhenium radioisotopes for medicine, a focus on production and applications. Molecules 2022, 27, 5283.
- Deutsch, E.; Libson, K.; Vanderheyden, J.L.; Ketring, A.R.; Maxon, H.R. The chemistry of rhenium and technetium as related to the use of isotopes of these elements in therapeutic and diagnostic nuclear medicine. Int. J. Rad. Appl. Instrum. B. 1986, 13, 465–477.
- Duatti, A. Fundamentals of technetium and rhenium chemistry. In Sampson’s Textbook of Radiopharmacy, 4th ed.; Theobald, T., Ed.; Pharmaceutical Press: London, UK, 2011; ISBN 10 0853697892/13 978-0853697893.
- Dilworth, J.R. Rhenium chemistry—Then and now. Coord. Chem. Rev. 2021, 436, 213822.
- Ferro-Flores, G.; de Murphy, C.A. Pharmacokinetics and dosimetry of 188Re-pharmaceuticals. Adv. Drug Deliv. Rev. 2008, 60, 1389–1401.
- Blower, P.J. A nuclear chocolate box: The periodic table of nuclear medicine. Dalton Trans. 2015, 44, 4819–4844.
- Knapp, F.F., Jr. Rhenium-188--a generator-derived radioisotope for cancer therapy. Cancer Biother. Radiopharm. 1998, 13, 337–349.
- Pillai, M.R.; Dash, A.; Knapp, F.F., Jr. Rhenium-188: Availability from the 188W/188Re generator and status of current applications. Curr. Radiopharm. 2012, 5, 228–243.
- Wunderlich, G.; Hartmann, H.; Andreeff, M.; Kotzerke, J. A semi-automated system for concentration of rhenium-188 for radiopharmaceutical applications. Appl. Radiat. Isot. 2008, 66, 1876–1880.
- Duatti, A. Review on 99mTc radiopharmaceuticals with emphasis on new advancements. Nucl. Med. Biol. 2021, 92, 202–216.
- Melis, D.R.; Burgoyne, A.R.; Ooms, M.; Gasser, G. Bifunctional chelators for radiorhenium: Past, present and future outlook. RSC Med. Chem. 2022, 13, 217–245.
- Bolzati, C.; Carta, D.; Salvarese, N.; Refosco, F. Chelating Systems for 99mTc/188Re in the development of radiolabeled peptide pharmaceuticals. Anti Cancer Agents Med. Chem. 2012, 12, 428–464.
- Shinto, A.; Knapp, F.F. Important clinical applications of 188Rhenium for radionuclide therapy. Int. J. Nucl. Medi. Res. Spec. Issue 2017, 16–38.
- Lepareur, N.; Ardisson, V.; Noiret, N.; Garin, E. (188)Re-SSS/Lipiodol: Development of a potential treatment for HCC from bench to bedside. Int. J. Mol. Imaging 2012, 2012, 278306.
- Ter Heine, R.; Lange, R.; Breukels, O.B.; Bloemendal, H.J.; Rummenie, R.G.; Wakker, A.M.; de Graaf, H.; Beekman, F.J.; van der Westerlaken, M.M.; Malingré, M.M.; et al. Bench to bedside development of GMP grade Rhenium-188-HEDP, a radiopharmaceutical for targeted treatment of painful bone metastases. Int. J. Pharm. 2014, 465, 317–324.
- Liepe, K.; Kropp, J.; Runge, R.; Kotzerke, J. Therapeutic efficiency of rhenium-188-HEDP in human prostate cancer skeletal metastases. Br. J. Cancer 2003, 89, 625–629.
- Lebuis, A.-M.; Beauchamp, A.L. Stereoisomerism of oxotrichloro(triphenylphosphine)rhenium(V), ReOCl3(PPh3)2: A reinvestigation. Can. J. Chem. 1993, 71, 441–449.
- Rouschias, G. Recent advances in the chemistry of rhenium. Chem. Rev. 1974, 74, 531–566.
- Kremer, C.; Rivero, M.; Kremer, E.; Suescun, L.; Mombrú, A.W.; Mariezcurrena, R.; Domínguez, S.; Mederos, A.; Midollini, S.; Castiñeiras, A. Synthesis, characterization and crystal structures of rhenium(V) complexes with diphosphines. Inorg. Chim. Acta 1999, 294, 47–55.
- Blake, A.J.; Greig, J.A.; Schröder, M. Rhenium complexes of tetra-aza macrocycles: The synthesis and single-crystal X-ray structure of trans-Cl·2(BPh3·H2O). J. Chem. Soc. Dalton Trans. 1988, 2645–2647.
- Abram, U.; Braun, M.; Abram, S.; Kirmse, R.; Voigt, A. : Facile synthesis, structure, electron paramagnetic resonance spectroscopy and reactions. J. Chem. Soc. Dalton Trans. 1998, 231–238.
- Boschi, A.; Bolzati, C.; Uccelli, L.; Duatti, A. High-yield synthesis of the terminal 188Re≡N multiple bond from generator-produced −. Nucl. Med. Biol. 2003, 30, 381–387.
- Corfield, P.W.R.; Doedens, R.J.; Ibers, A.J. Studies of metal-nitrogen multiple bonds. II. Crystal and molecular structure of nitridodichlorobistriphenylphosphine)rhenium(V), ReNCl2(P(C6H5)3)2. Inorg. Chem. 1967, 6, 204–210.
- Thieme, S.; Agostini, S.; Bergmann, R.; Pietzsch, J.; Pietzsch, H.-J.; Carta, D.; Salvarese, N.; Refosco, F.; Bolzati, C. Synthesis, Characterization and Biological Evaluation of +/0 mixed-ligand complexes as prototypes for the development of 188Re(N)-based target-specific radiopharmaceuticals. Nucl. Med. Biol. 2011, 38, 399–415.
- Carta, D.; Jentschel, C.; Thieme, S.; Salvarese, N.; Morellato, N.; Refosco, F.; Ruzza, P.; Bergmann, R.; Pietzsch, H.-J.; Bolzati, C. Assessment of the best N3– donors in preparation of -Based (M = 99mTc-; 188Re) target-specific radiopharmaceuticals: Comparison among succinic dihydrazide (SDH), N-methyl-S-methyl dithiocarbazate (HDTCZ) and PEGylated N-methyl-S-methyl dithiocarbazate (HO2C-PEG600-DTCZ). Nucl. Med. Biol. 2014, 41, 570–581.
- Fletcher, S.R.; Rowbottom, J.F.; Skapski, A.C.; Wilkinson, G. X-Ray crystal structure of diethyldithiocarbamate complexes of rhenium(V) with oxo- and nitrido-ligands. Chem. Commun. 1970, 22, 1572–1573.
More