Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 3 by Sirius Huang.
Epigenetics describes molecular missing link pathways that could bridge the gap between the genetic background and environmental risk factors that contribute to the pathogenesis of pulmonary fibrosis. Specific epigenetic patterns, especially DNA methylation, histone modifications, long non-coding, and microRNA (miRNAs), affect the endophenotypes underlying the development of idiopathic pulmonary fibrosis (IPF).
- epigenetics
- epigenomics
- DNA methylation
- long noncoding
- microRNA
- histone modification
- idiopathic pulmonary fibrosis
- chronic pulmonary diseases
1. Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, devastating, and irreversible lung disease that is characterized by microinjury-induced alveolar epithelial cell stress, progressive pathogenic myofibroblast differentiation, imbalanced macrophage polarization, and the extensive deposition of the extracellular matrix (ECM) [1][2][3]. The progression of patients with IPF is associated with lung function decline, progressive respiratory failure, high mortality, recurrent acute exacerbations, and an overall poor prognosis [4][5][6]. The morphological hallmark of IPF on histopathological and/or radiological is usual interstitial pneumonia (UIP), composed of heterogeneous areas of normal-appearing lung intermixed with collagenized fibrosis in sub-pleural and paraseptal, a honeycombing pattern, and ECM-producing myofibroblasts termed fibroblast foci (FF) [7][8].
A recent hypothesis stated that recurrent injuries drive the aberrant activation of epithelial cells to transdifferentiate into fibroblast epithelial-mesenchymal transition (EMT), which might induce fibrosis independently of inflammatory events [9][10]. Even though there is no implicit mechanism, several shreds of evidence emphasize that alveolar epithelial injury induced by environmental triggers results in lung fibrosis. Recurrent microenvironment injury on senescent epithelial cells in genetically susceptible individuals leads to the aberrant activation of fibroblasts, accumulating ECM, and fibrosis [11].
The pathogenic mechanisms involved in the initiation, development, and progression of IPF are unclear. However, many studies have demonstrated that dynamic interactions of genetic susceptibility, environmental factors, and host risk factors in older individuals contribute to epigenetic pro-fibrotic reprogramming, resulting in the development of IPF [12]. Hey et al. found a strong association between the microenvironment-driven epigenetic changes that could induce macrophage inflammation and polarization [13].
Omics-based approaches, including high-throughput technologies that provide snapshots of a holistic view of the molecules that make up a cell, tissue, or organism, consist of (1) genomics, measuring deoxyribonucleic acid (DNA) sequence variation; (2) epigenomics, focusing on the genome-wide characterization of reversible modifications of DNA or DNA alterations; (3) transcriptomics, evaluating the standard of ribonucleic acid (RNA) expression; (4) proteomics, determining protein expression or its chemical changes; (5) metabolomics, assessing metabolite/small molecule levels; and (6) microbiomics, investigating all the microorganisms of a given community [14][15]. Pulmonary disease omics studies mainly focus on tissue- and cell-specific omics data and have identified several fundamental mechanisms that underlie pulmonary biological processes, disease endotypes, and appropriate novel therapeutics for selected individuals [16].
Epigenomics is a technique for analyzing gene expression through epigenetic mechanisms, including DNA methylation, RNA, and histone modification [17]. These components interact and stabilize each other; therefore, the disruption of epigenetic nucleosomes can lead to their inappropriate expression, resulting in epigenetic disorders [18]. Epigenetics and epigenomics help explain how our environment affects our phenotype. Epigenomics studies commonly use methods such as Hi-C, a comprehensive technique developed to capture chromosome conformation, and another tool for whole genome methylation profiling: MBD-isolated genome sequencing (MiGS) [19]. Essential technical and experimental parameters that should be considered when designing epigenomic experiments are reviewed in detail by these authors [20].
2. DNA Methylation in Fibrosis
Extensive alterations in DNA methylation profiles are known to be involved in the pathogenesis of pulmonary fibrosis. DNA methylation microarray demonstrated a higher DNA methyltransferase expression in lung tissue samples of IPF patients [21]. IPF fibroblasts exhibited significant heterogeneity in global DNA methylation patterns compared to non-fibrotic control cells [22]. Additionally, a study identified extensive alterations in gene expression-associated DNA methylation that were involved in fibroproliferation [23]. Recently, a genome-wide DNA methylation study found that CpGs methylation was responsible for cell adhesion, molecule binding, chemical homeostasis, surfactant homeostasis, and receptor binding categories in the pathogenesis of IPF [24]. Global methylation patterns in IPF are very different from that of the control samples and significantly overlap with methylation changes observed in lung adenocarcinoma samples [25]. DNA methylation biomarkers also overlap between IPF, other ILD, cancer, and COPD; hence, it is unlikely that molecular discrimination between these diseases can be achieved using a single marker [26]. Interestingly, low-methylation lung squamous cell carcinoma (SCC) significantly correlated with IPF to show a bad outcome [27]. Cigarette smoking-associated aberrant methylation contributed to pulmonary fibrosis [28]. Many cell types are well known to be involved in the process of pulmonary fibrosis. The essential cells driving the development and progression of pulmonary fibrosis are epithelial, fibroblasts and myofibroblasts, and alveolar macrophages. Epigenetic regulation has a variety of ways. This discussion focuses on DNA methylation regulation in fibroblasts, epithelial, and macrophages.2.1. DNA Methylation and EMT
EMT is a cellular and molecular process in which epithelial cells lose their epithelial identity (apical-basal polarity and adhesion), which is characterized by down-regulated epithelial markers, including E-cadherin, occludin, and claudin-1. In contrast, fibroblast-specific genes, such as α-SMA, N-cadherin, fibroblast-specific protein 1 (FSP-1), and type I collagen, are up-regulated [10][29]. A hallmark of EMT is the repression of E-cadherin: a transmembrane glycoprotein encoded by the epithelial marker gene CDH1. DNA methylation might be involved in regulating EMT. During EMT induction, EMT-transcriptional factors (EMT-TFs), including ZEB1, SNAIL, and TWIST, interact with DNMTs and undergo the hypermethylation of CpG islands in the CDH1 promoter [30][31]. The DNA hypermethylation of the CDH1 through DNMTs was correlated with tumor progression [30][32]. In addition, the knockdown of DNMTs was equated with the transfection of siDNMT1, DNMT3a, or DNMT3b, which reversed the TGF-β1-induced suppression of E-cadherin expression and the induction of α-SMA, vimentin, and fibronectin expression [33]. In normal lung epithelial wound healing, this process ends with the apoptosis of myofibroblasts and inflammation reduction. Yet, aberrant responses to tissue injury may turn into lung fibrosis. Bidirectional EMT cross-talk assists the pro-fibrogenic positive feedback loop, while epithelial cells become “vulnerable and sensitive to apoptosis” and myofibroblasts become “apoptosis-resistant and immortal”, resulting in fibrosis progression instead of wound resolution [34]. DNA methylation mediated the down-regulation of genes involved in apoptosis. The hypermethylation of the Caspase 8 promoter by DNMT1 and DNMT3b was associated with apoptosis resistance in cancer cells [35][36]. Cisneros et al. showed the silencing expression of pro-apoptotic p14ARF in the fibroblast of the patient with IPF due to hypermethylation [37]. Along with these changes, DNA methylation was linked with the dysregulation of apoptosis and EMT, leading to the development of lung fibrosis processes (Figure 12).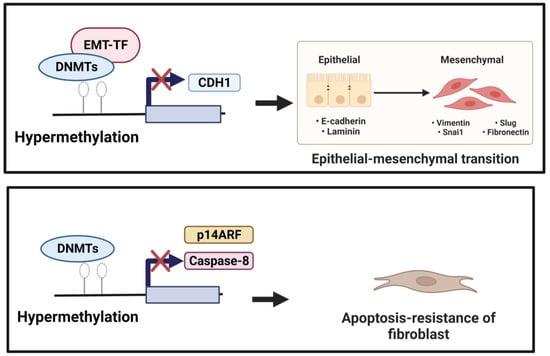
Figure 12. (1) Induction of EMT. Down-regulation of CDH1 (anti-fibrotic factor) is mediated by EMT-TF, which recruits DNMTs to the CDH1 promoter performing focal hypermethylation of the CpG islands in the CDH1 promoter, leading to decreased gene expression of CDH1. (2) Reduction in apoptotic activity. Hypermethylation mediated by DNMTs of the pro-apoptotic factors, including Caspase 8 and p14 ARF, could underlie resistance to fibroblast apoptosis.
2.2. DNA Methylation and Myofibroblast Differentiation
Under normal physiology, in response to epithelial injury, the cell releases various pro-inflammation and fibrotic cytokines that are responsible for local inflammation and the activation of fibroblasts. Fibroblasts are mesenchymal cells that are recruited to and accumulate at the injured site and undergo a change in phenotype to highly proliferative and contractile myofibroblasts. Lung myofibroblasts are heterogeneous in terms of their origins. The predominant sources of myofibroblasts are resident fibroblasts (lipofibroblasts, matrix fibroblasts, and alveolar niche cells) and pericytes (residing within basal membranes or perivascular linings), along with minor sources such as hematopoietic CXCR4+ fibrocytes, alveolar epithelial cells (AECs), endothelial cells (ECs), and mesenchymal stem cells (MSCs) [38]. Under the influence of environment-specific factors, cytokines, such as TGF-β, fibroblast-to-myofibroblast transition/transdifferentiation (FMT) changes lung resident fibroblast behavior/phenotypes to another type of normal differentiated cell and the downstream effects at the tissue level [39]. Myofibroblasts, characterized by the expression of contractile α-SMA, regulate connective tissue remodeling by producing and modifying ECM components, such as fibronectin and collagen [40]. Furthermore, one of the mechanisms that may account for FMT is DNA methylation. DNA methylation modulates myofibroblast differentiation (Figure 23). Several genes and signaling pathways, such as those involving ErbB, focal adhesion, and MAPK, underlie mechanisms for DNA methylation alteration in myofibroblast differentiation and influence the synthesis of ECM [41]. DNA methylation regulates gene expression to facilitate the formation of fibroblastic foci and lung fibrosis [42]. DNMT is an essential regulator of α-SMA gene expression during myofibroblast differentiation. The specific effect of DNA methylation in regulating the α-SMA gene is unknown. During the development of cardiac fibrosis, He et al. found that TGF-β could inhibit DNMT1-mediated DNA methylation expression, leading to the overexpression of α-SMA [43]. TGF-β1 also regulates lung fibroblast differentiation through the hypermethylation of the “fibrosis suppressor” gene, Thymocyte differentiation antigen-1 (THY-1); therefore, DNMT1-attenuated TGF1-mediated THY-1-inducing α-SMA fiber formation is silenced [44]. In addition, the expression of α-SMA was significantly increased by adding IL-6 [45]. A recent study revealed that DNMTl knockdown suppressed the DNA methylation-related myofibroblast phenotype via inhibiting an IL-6/STAT3/NF-κβ positive feedback loop [46].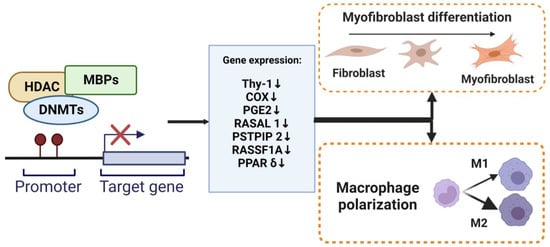
Figure 23. DNA methylation regulate myofibroblasts and macrophages. The methylation of promoter DNA can repress gene expression by MBPs, recruiting specific macromolecular complexes that contain histone deacetylases (HDAC), DNMTs, and other transcriptional co-repressors (Co-Rep). Several genes that function as an anti-fibrotic and are associated with myofibroblast differentiation and macrophage M2 polarization have been shown to undergo silencing due to DNA hypermethylation.
2.3. DNA Methylation and Macrophage Polarization
Pulmonary macrophages consist of monocyte-derived alveolar (AMs) and lung tissue-resident alveolar or interstitial macrophages (IMs). Both lung tissue-resident macrophages and monocyte-derived macrophages can be polarized into classically activated macrophages (M1) or alternatively activated macrophages (M2) by their capacity to induce inflammatory or anti-inflammatory immune responses, respectively [58]. The heterogeneity of macrophages regulates the development of pulmonary fibrosis from the early phases of injury and the fibrotic phase. The M2 macrophage, instead of the M1 phenotype, is involved in the progression of lung fibrosis [59]. After the resolution of the lung injury, monocyte-derived AM persisted and expressed higher pro-inflammatory and pro-fibrotic functions, and therefore, leading to the depletion of monocyte-derived AM ameliorates lung fibrosis [60]. DNA methylation alterations in IPF lungs have been well documented, but the role of DNA methylation in macrophages needs to be better defined. Many studies have shown that DNA methylation plays a fundamental role in macrophages, including the regulation of injury-induced inflammation and the control of fibroblast activation in the wound-healing response. Chen et al. examined DNA methylation changes in lung macrophages and found that changes in DNA methylation patterns drove the dysregulation of innate immune cells in cystic fibrosis [61]. Profiling DNA methylation from healthy subjects and IPF patients showed that aberrant macrophage polarization played a crucial role in developing IPF, but epigenetic alterations were not associated with accelerated aging [62]. DNMTs are associated with the activation of gene expression during macrophage phenotypic changes. During the development of IPF, DNMTs are associated with inappropriate gene expression activation, resulting in the opposite roles of M1/M2 polarization. DNMTs can act as pro- and anti-fibrotic by regulating M2 polarization. Yang et al. revealed that DNMT3a and DNMT3b suppress the Proline-Serine-Threonine Phosphatase Interacting Protein 2 (PSTPIP-2) by hypermethylation results, which caused a mixed induction of hepatic macrophage M1 and M2 [63]. In obesity, DNMT3b regulates M2 macrophage polarization, and the deletion of DNMT3b induces M2 macrophage polarization [64]. On the contrary, another study provided different results. Qin et al. found that DNMT3b ameliorates the development of bleomycin-induced pulmonary fibrosis by limiting M2 macrophage polarization [65]. In this case, DNMT3b acts as a negative regulator of M2 macrophage polarization. The role of DNMT3-associated macrophage polarization in organ fibrosis might have different functions. DNMT3 limits M2 macrophage polarization and alleviates the progression of lung fibrosis, yet DNMT3 induces M2 polarization and enhances liver fibrogenesis. Consistent with the involvement of DNMTs in macrophage phenotype change, MBPs interact with methylated DNA to regulate the expression of multiple genes. However, MeCP2 can accelerate both M1 and M2 polarization. MeCP2 might tend to M1 in the early phase of inflammation. The deletion of MeCP2 down-regulated M1 differentiation and inhibited the expression of iNOS and the secretion of cytokines that are pro-inflammatory in acute lung injury (ALI) [66]. MeCP2 regulates the gene expression of macrophages; thus, MeCP2 deficiency leads to the dysregulation of macrophage polarization [67]. MeCP2 accelerates M2 polarization by inhibiting Ship expression and enhancing the phosphatidylinositol 3-kinases/protein kinase B (PI3K/PKB) signaling pathway in bleomycin-induced pulmonary fibrosis [51]. Furthermore, silencing MeCP2 using siRNA blunted M2 macrophage polarization to elevate IRF4 expression [68]. Conversely, MeCP2 directly or indirectly promoted the differentiation of M0 macrophages to M1 or M2 macrophages and the polarization of M2 to M1 macrophages [69]. Therefore, researchers might suggest that MeCP2 stimulates M2 macrophages to promote the progression of lung fibrosis but protect against renal fibrosis via M1 polarization. Little is known about the impact of TET proteins on macrophage polarization. TET regulates macrophage differentiation and M1-polarization [70]. Therefore, the inhibition of the TET protein using dimethyloxallyl glycine (DMOG) promotes M2 polarization by up-regulating Arg1, Fizz1, and Ym1 [71].References
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32.
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018, 27, 148.
- Kishore, A.; Petrek, M. Roles of Macrophage Polarization and Macrophage-Derived miRNAs in Pulmonary Fibrosis. Front. Immunol. 2021, 12, 678457.
- Fernández Fabrellas, E.; Peris Sánchez, R.; Sabater Abad, C.; Juan Samper, G. Prognosis and Follow-Up of Idiopathic Pulmonary Fibrosis. Med. Sci. 2018, 6, 51.
- Alqalyoobi, S.; Fernández Pérez, E.R.; Oldham, J.M. In-hospital mortality trends among patients with idiopathic pulmonary fibrosis in the United States between 2013–2017: A comparison of academic and non-academic programs. BMC Pulm. Med. 2020, 20, 289.
- Yoo, J.-W.; Kim, J.; Song, J.W. Impact of the revised definition on incidence and outcomes of acute exacerbation of idiopathic pulmonary fibrosis. Sci. Rep. 2022, 12, 8817.
- Aburto, M.; Herráez, I.; Iturbe, D.; Jiménez-Romero, A. Diagnosis of Idiopathic Pulmonary Fibrosis: Differential Diagnosis. Med. Sci. 2018, 6, 73.
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68.
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823.
- Lovisa, S. Epithelial-to-Mesenchymal Transition in Fibrosis: Concepts and Targeting Strategies. Front. Pharmacol. 2021, 12, 737570.
- Pardo, A.; Selman, M. Lung fibroblasts, aging, and idiopathic pulmonary fibrosis. Ann. Am. Thorac. Soc. 2016, 13, S417–S421.
- Pardo, A.; Selman, M. The Interplay of the Genetic Architecture, Aging, and Environmental Factors in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 64, 163–172.
- Hey, J.; Paulsen, M.; Toth, R.; Weichenhan, D.; Butz, S.; Schatterny, J.; Liebers, R.; Lutsik, P.; Plass, C.; Mall, M.A. Epigenetic reprogramming of airway macrophages promotes polarization and inflammation in muco-obstructive lung disease. Nat. Commun. 2021, 12, 6520.
- Horgan, R.P.; Kenny, L.C. ‘Omic’ technologies: Genomics, transcriptomics, proteomics and metabolomics. Obstet. Gynaecol. 2011, 13, 189–195.
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83.
- Kan, M.; Shumyatcher, M.; Himes, B.E. Using omics approaches to understand pulmonary diseases. Respir. Res. 2017, 18, 149.
- Wang, K.C.; Chang, H.Y. Epigenomics: Technologies and Applications. Circ. Res. 2018, 122, 1191–1199.
- Guiot, J.; Struman, I.; Chavez, V.; Henket, M.; Herzog, M.; Scoubeau, K.; Hardat, N.; Bondue, B.; Corhay, J.L.; Moermans, C.; et al. Altered epigenetic features in circulating nucleosomes in idiopathic pulmonary fibrosis. Clin. Epigenetics 2017, 9, 84.
- Dai, X.; Shen, L. Advances and Trends in Omics Technology Development. Front. Med. 2022, 9, 1546.
- Mehrmohamadi, M.; Sepehri, M.H.; Nazer, N.; Norouzi, M.R. A Comparative Overview of Epigenomic Profiling Methods. Front. Cell Dev. Biol. 2021, 9, 714687.
- Sanders, Y.Y.; Ambalavanan, N.; Halloran, B.; Zhang, X.; Liu, H.; Crossman, D.K.; Bray, M.; Zhang, K.; Thannickal, V.J.; Hagood, J.S. Altered DNA Methylation Profile in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 525–535.
- Huang, S.K.; Scruggs, A.M.; McEachin, R.C.; White, E.S.; Peters-Golden, M. Lung Fibroblasts from Patients with Idiopathic Pulmonary Fibrosis Exhibit Genome-Wide Differences in DNA Methylation Compared to Fibroblasts from Nonfibrotic Lung. PLoS ONE 2014, 9, e107055.
- Yang, I.V.; Pedersen, B.S.; Rabinovich, E.; Hennessy, C.E.; Davidson, E.J.; Murphy, E.; Guardela, B.J.; Tedrow, J.R.; Zhang, Y.; Singh, M.K.; et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1263–1272.
- Lee, J.-U.; Son, J.-H.; Shim, E.-Y.; Cheong, H.S.; Shin, S.-W.; Shin, H.D.; Baek, A.R.; Ryu, S.; Park, C.-S.; Chang, H.S.; et al. Global DNA Methylation Pattern of Fibroblasts in Idiopathic Pulmonary Fibrosis. DNA Cell Biol. 2019, 38, 905–914.
- Rabinovich, E.I.; Kapetanaki, M.G.; Steinfeld, I.; Gibson, K.F.; Pandit, K.V.; Yu, G.; Yakhini, Z.; Kaminski, N. Global Methylation Patterns in Idiopathic Pulmonary Fibrosis. PLoS ONE 2012, 7, e33770.
- Wielscher, M.; Vierlinger, K.; Kegler, U.; Ziesche, R.; Gsur, A.; Weinhäusel, A. Diagnostic Performance of Plasma DNA Methylation Profiles in Lung Cancer, Pulmonary Fibrosis and COPD. EBioMedicine 2015, 2, 929–936.
- Hata, A.; Nakajima, T.; Matsusaka, K.; Fukuyo, M.; Morimoto, J.; Yamamoto, T.; Sakairi, Y.; Rahmutulla, B.; Ota, S.; Wada, H.; et al. A low DNA methylation epigenotype in lung squamous cell carcinoma and its association with idiopathic pulmonary fibrosis and poorer prognosis. Int. J. Cancer 2020, 146, 388–399.
- de Vries, M.; van der Plaat, D.A.; Nedeljkovic, I.; Verkaik-Schakel, R.N.; Kooistra, W.; Amin, N.; van Duijn, C.M.; Brandsma, C.-A.; van Diemen, C.C.; Vonk, J.M.; et al. From blood to lung tissue: Effect of cigarette smoke on DNA methylation and lung function. Respir. Res. 2018, 19, 212.
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483.
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18.
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-Mesenchymal Transition: Epigenetic Reprogramming Driving Cellular Plasticity. Trends Genet. 2017, 33, 943–959.
- Urbanova, M.; Buocikova, V.; Trnkova, L.; Strapcova, S.; Kajabova, V.H.; Melian, E.B.; Novisedlakova, M.; Tomas, M.; Dubovan, P.; Earl, J.; et al. DNA Methylation Mediates EMT Gene Expression in Human Pancreatic Ductal Adenocarcinoma Cell Lines. Int. J. Mol. Sci. 2022, 23, 2117.
- Park, J.-H.; Shin, J.-M.; Yang, H.-W.; Park, I.-H. DNMTs Are Involved in TGF-β1-Induced Epithelial-Mesenchymal Transitions in Airway Epithelial Cells. Int. J. Mol. Sci. 2022, 23, 3003.
- Yao, L.; Zhou, Y.; Li, J.; Wickens, L.; Conforti, F.; Rattu, A.; Ibrahim, F.M.; Alzetani, A.; Marshall, B.G.; Fletcher, S.V.; et al. Bidirectional epithelial-mesenchymal crosstalk provides self-sustaining profibrotic signals in pulmonary fibrosis. J. Biol. Chem. 2021, 297, 101096.
- Hervouet, E.; Cheray, M.; Vallette, F.M.; Cartron, P.-F. DNA Methylation and Apoptosis Resistance in Cancer Cells. Cells 2013, 2, 545–573.
- Kostova, I.; Mandal, R.; Becker, S.; Strebhardt, K. The role of caspase-8 in the tumor microenvironment of ovarian cancer. Cancer Metastasis Rev. 2021, 40, 303–318.
- Cisneros, J.; Hagood, J.; Checa, M.; Ortiz-Quintero, B.; Negreros, M.; Herrera, I.; Ramos, C.; Pardo, A.; Selman, M. Hypermethylation-mediated silencing of p14(ARF) in fibroblasts from idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L295–L303.
- Lai, Y.; Wei, X.; Ye, T.; Hang, L.; Mou, L.; Su, J. Interrelation Between Fibroblasts and T Cells in Fibrosing Interstitial Lung Diseases. Front. Immunol. 2021, 12, 747335.
- D’Urso, M.; Kurniawan, N.A. Mechanical and Physical Regulation of Fibroblast–Myofibroblast Transition: From Cellular Mechanoresponse to Tissue Pathology. Front. Bioeng. Biotechnol. 2020, 8, 1459.
- Doolin, M.T.; Smith, I.M.; Stroka, K.M. Fibroblast to myofibroblast transition is enhanced by increased cell density. Mol. Biol. Cell 2021, 32, ar41.
- Li, J.; Yao, W.; Zhang, L.; Bao, L.; Chen, H.; Wang, D.; Yue, Z.; Li, Y.; Zhang, M.; Hao, C. Genome-wide DNA methylation analysis in lung fibroblasts co-cultured with silica-exposed alveolar macrophages. Respir. Res. 2017, 18, 91.
- Luo, Q.-K.; Zhang, H.; Li, L. Research Advances on DNA Methylation in Idiopathic Pulmonary Fibrosis. In Single-Cell Sequencing and Methylation: Methods and Clinical Applications; Yu, B., Zhang, J., Zeng, Y., Li, L., Wang, X., Eds.; Springer: Singapore, 2020; pp. 73–81. ISBN 978-981-15-4494-1.
- He, Y.; Ling, S.; Sun, Y.; Sheng, Z.; Chen, Z.; Pan, X.; Ma, G. DNA methylation regulates α-smooth muscle actin expression during cardiac fibroblast differentiation. J. Cell. Physiol. 2019, 234, 7174–7185.
- Neveu, W.A.; Mills, S.T.; Staitieh, B.S.; Sueblinvong, V. TGF-β1 epigenetically modifies Thy-1 expression in primary lung fibroblasts. Am. J. Physiol. Cell Physiol. 2015, 309, C616–C626.
- Ogawa, H.; Koyanagi-Aoi, M.; Otani, K.; Zen, Y.; Maniwa, Y.; Aoi, T. Interleukin-6 blockade attenuates lung cancer tissue construction integrated by cancer stem cells. Sci. Rep. 2017, 7, 12317.
- Al-Kharashi, L.A.; Al-Mohanna, F.H.; Tulbah, A.; Aboussekhra, A. The DNA methyl-transferase protein DNMT1 enhances tumor-promoting properties of breast stromal fibroblasts. Oncotarget 2018, 9, 2329–2343.
- Evans, I.C.; Barnes, J.L.; Garner, I.M.; Pearce, D.R.; Maher, T.M.; Shiwen, X.; Renzoni, E.A.; Wells, A.U.; Denton, C.P.; Laurent, G.J.; et al. Epigenetic regulation of cyclooxygenase-2 by methylation of c8orf4 in pulmonary fibrosis. Clin. Sci. 2016, 130, 575–586.
- Mann, J.; Oakley, F.; Akiboye, F.; Elsharkawy, A.; Thorne, A.W.; Mann, D.A. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: Implications for wound healing and fibrogenesis. Cell Death Differ. 2007, 14, 275–285.
- Xiang, Z.; Zhou, Q.; Hu, M.; Sanders, Y.Y. MeCP2 epigenetically regulates alpha-smooth muscle actin in human lung fibroblasts. J. Cell. Biochem. 2020, 121, 3616–3625.
- Shin, J.-M.; Um, J.-Y.; Lee, S.-A.; Park, I.-H.; Lee, S.-H.; Lee, H.-M. Effect of MeCP2 on TGF-β1-induced Extracellular Matrix Production in Nasal Polyp-derived Fibroblasts. Am. J. Rhinol. Allergy 2018, 32, 228–235.
- Wang, Y.; Zhang, L.; Wu, G.-R.; Zhou, Q.; Yue, H.; Rao, L.-Z.; Yuan, T.; Mo, B.; Wang, F.-X.; Chen, L.-M.; et al. MBD2 serves as a viable target against pulmonary fibrosis by inhibiting macrophage M2 program. Sci. Adv. 2021, 7, eabb6075.
- Wang, Y.; Zhang, L.; Huang, T.; Wu, G.-R.; Zhou, Q.; Wang, F.-X.; Chen, L.-M.; Sun, F.; Lv, Y.; Xiong, F.; et al. The methyl-CpG-binding domain 2 facilitates pulmonary fibrosis by orchestrating fibroblast to myofibroblast differentiation. Eur. Respir. J. 2022, 60, 2003697.
- He, Y.; Tsou, P.-S.; Khanna, D.; Sawalha, A.H. Methyl-CpG-binding protein 2 mediates antifibrotic effects in scleroderma fibroblasts. Ann. Rheum. Dis. 2018, 77, 1208–1218.
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.T.; McDonald, K.M.; McCann, A.; et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum. Mol. Genet. 2014, 23, 2176–2188.
- Robinson, C.M.; Neary, R.; Levendale, A.; Watson, C.J.; Baugh, J.A. Hypoxia-induced DNA hypermethylation in human pulmonary fibroblasts is associated with Thy-1 promoter methylation and the development of a pro-fibrotic phenotype. Respir. Res. 2012, 13, 74.
- McDonnell, F.; Irnaten, M.; Clark, A.F.; O’Brien, C.J.; Wallace, D.M. Hypoxia-Induced Changes in DNA Methylation Alter RASAL1 and TGFβ1 Expression in Human Trabecular Meshwork Cells. PLoS ONE 2016, 11, e0153354.
- Rajgarhia, A.; Ayasolla, K.R.; Zaghloul, N.; Lopez Da Re, J.M.; Miller, E.J.; Ahmed, M. Extracellular Superoxide Dismutase (EC-SOD) Regulates Gene Methylation and Cardiac Fibrosis During Chronic Hypoxic Stress. Front. Cardiovasc. Med. 2021, 8, 669975.
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.-Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 170.
- Cheng, P.; Li, S.; Chen, H. Macrophages in Lung Injury, Repair, and Fibrosis. Cells 2021, 10, 436.
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.-I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404.
- Chen, Y.; Armstrong, D.A.; Salas, L.A.; Hazlett, H.F.; Nymon, A.B.; Dessaint, J.A.; Aridgides, D.S.; Mellinger, D.L.; Liu, X.; Christensen, B.C.; et al. Genome-wide DNA methylation profiling shows a distinct epigenetic signature associated with lung macrophages in cystic fibrosis. Clin. Epigenetics 2018, 10, 152.
- McErlean, P.; Bell, C.G.; Hewitt, R.J.; Busharat, Z.; Ogger, P.P.; Ghai, P.; Albers, G.J.; Calamita, E.; Kingston, S.; Molyneaux, P.L.; et al. DNA Methylome Alterations Are Associated with Airway Macrophage Differentiation and Phenotype during Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 204, 954–966.
- Yang, Y.; Wu, X.; Li, W.; Huang, H.; Li, H.; Pan, X.; Li, X.; Huang, C.; Meng, X.; Zhang, L.; et al. PSTPIP2 connects DNA methylation to macrophage polarization in CCL4-induced mouse model of hepatic fibrosis. Oncogene 2018, 37, 6119–6135.
- Wang, X.; Cao, Q.; Yu, L.; Shi, H.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight 2016, 1, e87748.
- Qin, W.; Spek, C.A.; Scicluna, B.P.; van der Poll, T.; Duitman, J. Myeloid DNA methyltransferase3b deficiency aggravates pulmonary fibrosis by enhancing profibrotic macrophage activation. Respir. Res. 2022, 23, 162.
- Sun, W. The role of MBD2 regulating M1 macrophage polarization in acute lung injury. Eur. Respir. J. 2022, 60, 4589.
- Zhao, D.; Mokhtari, R.; Pedrosa, E.; Birnbaum, R.; Zheng, D.; Lachman, H.M. Transcriptome analysis of microglia in a mouse model of Rett syndrome: Differential expression of genes associated with microglia/macrophage activation and cellular stress. Mol. Autism 2017, 8, 17.
- Mou, Y.; Wu, G.-R.; Wang, Q.; Pan, T.; Zhang, L.; Xu, Y.; Xiong, W.; Zhou, Q.; Wang, Y. Macrophage-targeted delivery of siRNA to silence Mecp2 gene expression attenuates pulmonary fibrosis. Bioeng. Transl. Med. 2022, 7, e10280.
- Ai, K.; Pan, J.; Zhang, P.; Li, H.; He, Z.; Zhang, H.; Li, X.; Li, Y.; Yi, L.; Kang, Y.; et al. Methyl-CpG-binding domain protein 2 contributes to renal fibrosis through promoting polarized M1 macrophages. Cell Death Dis. 2022, 13, 125.
- Cull, A.; Snetsinger, B.; Rauh, M.J. Tet2 Is a Novel Regulator of Murine Macrophage Differentiation and Polarization. Blood 2015, 126, 646.
- Hams, E.; Saunders, S.P.; Cummins, E.P.; O’Connor, A.; Tambuwala, M.T.; Gallagher, W.M.; Byrne, A.; Campos-Torres, A.; Moynagh, P.M.; Jobin, C.; et al. The hydroxylase inhibitor dimethyloxallyl glycine attenuates endotoxic shock via alternative activation of macrophages and IL-10 production by B1 cells. Shock 2011, 36, 295–302.
More